
A Unified Method for Detecting Secondary Trait Associations with Rare Variants: Application to Sequence Data
Next-generation sequencing has made possible the detection of rare variant (RV) associations with quantitative traits (QT). Due to high sequencing cost, many studies can only sequence a modest number of selected samples with extreme QT. Therefore association testing in individual studies can be underpowered. Besides the primary trait, many clinically important secondary traits are often measured. It is highly beneficial if multiple studies can be jointly analyzed for detecting associations with commonly measured traits. However, analyzing secondary traits in selected samples can be biased if sample ascertainment is not properly modeled. Some methods exist for analyzing secondary traits in selected samples, where some burden tests can be implemented. However p-values can only be evaluated analytically via asymptotic approximations, which may not be accurate. Additionally, potentially more powerful sequence kernel association tests, variable selection-based methods, and burden tests that require permutations cannot be incorporated. To overcome these limitations, we developed a unified method for analyzing secondary trait associations with RVs (STAR) in selected samples, incorporating all RV tests. Statistical significance can be evaluated either through permutations or analytically. STAR makes it possible to apply more powerful RV tests to analyze secondary trait associations. It also enables jointly analyzing multiple cohorts ascertained under different study designs, which greatly boosts power. The performance of STAR and commonly used RV association tests were comprehensively evaluated using simulation studies. STAR was also implemented to analyze a dataset from the SardiNIA project where samples with extreme low-density lipoprotein levels were sequenced. A significant association between LDLR and systolic blood pressure was identified, which is supported by pharmacogenetic studies. In summary, for sequencing studies, STAR is an important tool for detecting secondary-trait RV associations.
Published in the journal:
. PLoS Genet 8(11): e32767. doi:10.1371/journal.pgen.1003075
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003075
Summary
Next-generation sequencing has made possible the detection of rare variant (RV) associations with quantitative traits (QT). Due to high sequencing cost, many studies can only sequence a modest number of selected samples with extreme QT. Therefore association testing in individual studies can be underpowered. Besides the primary trait, many clinically important secondary traits are often measured. It is highly beneficial if multiple studies can be jointly analyzed for detecting associations with commonly measured traits. However, analyzing secondary traits in selected samples can be biased if sample ascertainment is not properly modeled. Some methods exist for analyzing secondary traits in selected samples, where some burden tests can be implemented. However p-values can only be evaluated analytically via asymptotic approximations, which may not be accurate. Additionally, potentially more powerful sequence kernel association tests, variable selection-based methods, and burden tests that require permutations cannot be incorporated. To overcome these limitations, we developed a unified method for analyzing secondary trait associations with RVs (STAR) in selected samples, incorporating all RV tests. Statistical significance can be evaluated either through permutations or analytically. STAR makes it possible to apply more powerful RV tests to analyze secondary trait associations. It also enables jointly analyzing multiple cohorts ascertained under different study designs, which greatly boosts power. The performance of STAR and commonly used RV association tests were comprehensively evaluated using simulation studies. STAR was also implemented to analyze a dataset from the SardiNIA project where samples with extreme low-density lipoprotein levels were sequenced. A significant association between LDLR and systolic blood pressure was identified, which is supported by pharmacogenetic studies. In summary, for sequencing studies, STAR is an important tool for detecting secondary-trait RV associations.
Introduction
Next-generation sequencing has already revolutionized the study of complex traits, and made possible the detection of rare variant associations. Many sequence based association studies are currently being performed, some of which have already lead to the discovery of associations with clinically important traits, such as lipids levels [1], age related macular degeneration [2], inflammatory bowel disease [3], blood pressure [4], body mass index [5], etc. In particular, there is increasing interest to detect associations with quantitative traits (QT). It has been suggested that complex traits can be due to multiple variants with small effects, and are quantitative in nature [6], [7]. Mapping multiple quantitative traits may help elucidate the etiology of complex traits, reducing sample heterogeneity [8], dissecting gene pleiotropy and refine the definition of complex diseases [7], [9], [10]. For example, recent studies of type 2 diabetes have been focused on multiple related QTs, such as fasting glucose levels [11], insulin resistance levels [11], and c-reactive proteins [12]. Many quantitative traits are usually measured in different studies as secondary outcomes. It is of great interest to combine multiple studies for detecting associations with commonly measured primary or secondary traits. For example, the National Heart Lung and Blood Institute-Exome Sequencing Project (NHLBI-ESP) is studying a variety of different phenotypes and employed both extreme and random sampling. In order to improve power, all samples with the phenotype of interest measured are jointly analyzed. Specifically, the association analysis of low density lipoprotein cholesterol (LDL) levels is performed by combining several studies which include a well-phenotyped random population cohort, selected samples with extreme LDL levels as well as individuals with extreme body mass index (BMI).
Many methods have been developed for detecting rare variant associations [13]–[19]. These methods are all based upon aggregating multiple rare variants across a genetic region, which is usually a gene. Compared to analyzing each variant individually, these region based tests can be more powerful. However, rare variants that are involved in complex trait etiologies usually only have moderate effect sizes, and their aggregated frequencies across a genetic region can still be low. It is therefore necessary to sequence and analyze a large number of samples in order to have adequate power to detect associations. Although next generation sequencing is much more cost effective than Sanger sequencing, it is still expensive to generate high read depth data for large numbers of samples. Given cost constraints, in order to improve power, many studies sequence samples with extreme QT rather than the entire cohort. The selective sampling study design produces challenges for analyzing secondary traits. Without properly accounting for the sample ascertainment mechanisms, type I errors for detecting secondary trait associations may be inflated [20], [21]. This is because due to the correlations between the primary and secondary traits, mean values for the secondary traits will be different between individuals with primary trait from opposite extreme tails. Additionally if the primary trait is associated with a gene region, the cumulative variant frequencies will also be different. Therefore spurious association can be created by ascertainment. The bias for the naïve analysis of secondary trait is demonstrated both theoretically and empirically in this article.
Several methods exist for detecting secondary trait associations in selected samples. For example, a retrospective likelihood method was developed for mapping secondary phenotypes using regression models (SPREG) in case control studies [20]. It was subsequently extended in an empirical Bayesian framework [22], which utilizes genotype information from cases for rare diseases and can be more powerful than the retrospective likelihood method. However, both methods are not directly applicable to the studies where the primary trait is quantitative and extreme sampling is implemented. We previously developed a method for detecting multiple (secondary) trait associations (MTA) in selected samples, which jointly models multiple phenotype associations and sampling ascertainment status [21]. MTA can be used to analyze data from studies with known sampling mechanisms, e.g. case control, and extreme sampling designs. It incorporates several rare variant association tests, whose statistical significance can be evaluated analytically e.g. the combined multivariate and collapsing (CMC) [16], and Gene- or Region-based Analysis of Variants of Intermediate and Low frequency (GRANVIL) [18]. Weighted sum statistics [14], [23] can also be incorporated, if the weights that are assigned to each variant site are not dependent on the trait. One major advantage of using MTA is that cohorts ascertained under different sampling schemes can be combined for detecting associations with commonly measured traits. These studies can be targeted at the same or different primary traits. By combining data from different studies, much larger sample sizes can be analyzed and the power to detect associations can be greatly improved [21].
However none of these methods for detecting secondary associations incorporate sequence kernel association test (SKAT), a powerful variance component score test based method. This method can be more powerful when causal variants have bidirectional effects and/or a large proportion of the variants within gene region are non-causal.
Standard permutation algorithms cannot be applied to obtain empirical p-value. This is because when the primary and secondary traits are correlated and the genetic region is associated with the primary trait, neither the secondary trait residuals nor the locus genotypes are interchangeable under the null hypothesis. Therefore, the statistical significance can only be evaluated via asymptotic approximations, which has several notable limitations: 1.) Due to the low frequency of rare variants, asymptotic approximation for some tests may be violated, which can lead to either inflated type I error or loss of power. 2.) For some rare variant association methods, the analytical distribution for the test statistics is unknown and therefore the statistical significance has to be evaluated empirically. These rare variant tests that require evaluating p-values via permutation are often more powerful than the methods implemented in MTA, e.g. CMC or GRANVIL. It is therefore desirable that these tests can be applied to analyze secondary traits.
To overcome the limitations of existing methods, a unified model was developed to detect secondary trait associations using selected samples. In the samples with extreme primary quantitative traits, through re-parameterizing the likelihood functions, interchangeable residuals for the secondary traits can be obtained under the null hypothesis. The residuals are approximately independent, and normally distributed. We proved theoretically that the analysis of secondary trait associations can be equivalently implemented by analyzing the correlation between the secondary trait residuals and the gene/genetic region. Therefore, any rare variant association test that can analyze QT in random population based studies can be incorporated in STAR. In addition, multiple cohorts can be jointly analyzed through conventional mega-analysis methods that use individual participant data or meta-analysis methods that use summary level statistics.
A variety of popular rare variant tests have been implemented in the STAR framework and the power to detect secondary trait associations was evaluated. Specifically, we considered the weighted sum statistic (WSS) [14], [23], sequence kernel association tests (SKAT) [17], and variable threshold test (VT) [24]. Additionally the kernel based adaptive cluster test (KBAC) [15], which was originally developed for analyzing binary disorders, was extended to analyze quantitative traits and incorporated in STAR for detecting associations with secondary phenotypes (Text S1).
The performances for these methods were compared using extensive simulation studies. Genetic data were simulated under a realistic population genetic model as described by Kryukov et al [25], which incorporates both demographic change and purifying selections. Phenotypes were simulated based upon parameters estimated from clinically important complex traits. It is demonstrated that under a broad variety of phenotype models, the power for detecting secondary trait associations can be greatly improved through the use of more powerful rare variant tests that are incorporated in STAR. There does not exist a method that is consistently the most powerful, and the power difference between top performing methods is generally small. When the effects of causal variants are unidirectional, the VT test outperforms other methods in most scenarios. When there are variants with effects in opposite directions or only a small proportion of the variants are causal, SKAT can be more powerful than alternative methods in many settings.
The STAR method was also used to analyze a published sequence dataset from the SardiNIA project [1], where nine genes were sequenced in 256 individuals with extreme LDL levels (individuals taking lipid-lowering therapies were not considered for the analysis). In the original article by Sanna et al [1], the authors focused on detecting associations with the primary trait LDL, and did not consider analyzing other metabolic and lipids traits. In this article, the analysis was extended to detect associations with other clinically important traits, which include high density lipoprotein cholesterol (HDL), total cholesterol level (TCL), triglyceride (TG), insulin levels (INSULIN), BMI, systolic and diastolic blood pressure (SysBP and DiasBP). One association was identified between LDLR and SysBP, which is statistically significant after applying a Bonferroni correction for testing multiple genes and traits. This association has strong biological support from pharmacogenetics studies [26]. These findings provide new insight on the etiology for the LDLR gene, and established the importance of our method in sequence based association studies.
An R-package, STARSEQ which implements the STAR method is available through the Comprehensive R Archive Network (CRAN) at http://cran.r-project.org/web/packages/STARSEQ/. Additional companion softwares are deposited at http://code.google.com/p/starseq/.
Materials and Methods
STAR model can be used with any rare variant test to detect associations with secondary traits in studies that use extreme sampling. The multi-site genotype for individual is denoted by a vector , where is the total number of variant sites that are jointly analyzed. The set of variants can be determined by variant frequency threshold (either fixed or variable), or functional annotations (e.g. non-synonymous variants) etc. Each entry in the vector can be coded by the number of minor alleles, e.g. if the genotype is homozygous for the minor allele. The primary QT is denoted by , while the secondary trait under consideration is denoted by . In vector (or matrix) notations, the primary traits, secondary traits and genotypes for the entire sample are respectively denoted by , and .
Ascertainment Corrected Likelihood Model under the Null Hypothesis
Under the null hypothesis of no gene/secondary trait associations, following the MTA framework, a multivariate generalized linear model can be implemented to estimate nuisance parameters [21]. The link functions for the mean parameters of the two traits satisfy

The residual terms for the secondary traits, i.e. are correlated with the primary trait, and not interchangeable under the null hypothesis, i.e.

In order to obtain unbiased results, sampling schemes have to be properly modeled. Ascertainment corrected likelihood can be used, which jointly models sample ascertainment status and genotype/phenotype association, i.e.

Re-Parameterization of the Likelihood Function
The likelihood function in equation (1) needs to be re-parameterized in order to facilitate deriving the SKAT statistics and performing permutations. It is clear that



In this model, the residual errors and for the primary and secondary traits are uncorrelated. In particular, the residual errors after re-parameterization are interchangeable under the null hypothesis.
Score Statistics for Burden Tests and Variance Component Score Test
Burden tests, such as CMC and WSS, aggregate multiple rare variants across a genetic region and analyze them jointly. The following model can be used to obtain score statistics for a burden test:

Score tests can be formally constructed from the joint likelihood for testing the null hypothesis of no gene/secondary trait associations, i.e. . If the samples are ascertained based upon only the primary trait, score tests can be equivalently constructed from the conditional likelihood, i.e. . This is because the joint likelihood can be factorized, i.e.



It is clear from formula (9) that is proportional to the covariance between the secondary trait residuals and the locus genotype coding. Given that , , and are consistent estimators under the null hypothesis, by Slutsky's theorem, the residuals for the secondary trait i.e. are approximately normally distributed and interchangeable under the null hypothesis. Therefore, the analysis of rare variant secondary trait associations can be implemented by analyzing the correlation between the corrected residuals and the locus genotype coding. Standard permutation algorithms can be implemented by shuffling the residuals under the null hypothesis. In our STARSEQ package, we also provide flexible tools for calculating the adjusted secondary trait residuals, which can be analyzed by any user specified rare variant association test.
Using similar ideas, we show in (Text S5), that the extended SKAT statistic in STAR has the form

Extensions of KBAC Test to Analyze Quantitative Trait Associations
The KBAC test was previously developed for the analysis of binary trait associations [15]. It is extended to analyze rare variant QT associations in studies using randomly ascertained samples or samples with extreme traits. The extended KBAC method has also been incorporated in STAR for analyzing secondary trait associations. The details for the extensions are given in (Text S1).
Evaluation of Type I Errors and Power
Type I error and power were evaluated for the following rare variant association tests that were extended in STAR, i.e. CMC, KBAC, WSS, SKAT and VT. Genetic data were generated according to a four parameter demographic model for Europeans [25], [27]. In addition, purifying selection is also modeled, which influences the variant site frequency spectrum. Among the variants with selection coefficients >10−4, 50% are randomly chosen to be causal for the primary trait, and another 50% of the variants are independently chosen to be causal for the secondary trait. The set of causal variants for the primary trait are denoted by and that for the secondary trait are denoted by . Variants belonging to the intersection of and modulate both the primary and secondary phenotypes. QTs were simulated according to the following bivariate normal distribution:


Data for selective sampling studies are simulated, where for each dataset, a total of 5,000 individuals with extreme primary trait values are selected from a cohort of 100,000 individuals. Two-sided alternative hypothesis was tested for each method. Although p-values for CMC and SKAT can be obtained analytically, they can either be conservative or anti-conservative [23]. In order to calibrate the distribution of p-values, we evaluated the statistical significance of all methods empirically using 5,000 permutations. The power and type-I errors for each method were obtained using 10,000 replicates for a significance level of . As a comparison to STAR, type I error for linear regression analysis was also evaluated, where sample ascertainment mechanisms were ignored.
Application of STAR to Meta-Analysis
In order to illustrate the application of STAR for combining multiple cohorts, a meta-analysis of three studies was simulated. The primary trait for each study is different and a common secondary trait is measured for all studies. In the first study, the gene region is associated with the primary trait, and causal variants have an effect of −0.5. The correlation between the primary and secondary traits is 0.6. In the second study, the primary trait is associated with the gene region, and causal variants have an effect of 0.25. The primary and secondary traits are correlated with coefficient 0.4. In the third study, the gene region is not associated with the primary trait, and the correlation between the primary and secondary traits is −0.2. In each study, a different pool of 50,000 samples was simulated and 2,500 individuals with extreme primary trait were selected and analyzed for association. P-values for all rare variant tests in each study were obtained based upon 5,000 permutations. Meta-analysis is performed by combining Z-score statistics, which are transformed from p-values and weighted by the square root of the sample sizes in each study [28].
In order to evaluate type I errors, data were simulated under the assumption that the secondary trait effects for all variants were 0. The empirical distribution of p-values was obtained using 10,000 replicates. For evaluating power, two scenarios were considered, i.e. (A) causal variants have an unidirectional effect of 0.5; (B) causal variants have bidirectional effects, where 80% of the causal variants have effect 0.5 and the other 20% of the causal variants have effect −0.5.The power for analyzing each individual study and meta-analysis was evaluated using 10,000 replicates under a significance level of α = 0.05.
The Analysis of the SardiNIA Sequence Dataset
Association analyses were performed for the nine genes that were sequenced from the SardiNIA project [1]. First, coding regions of four genes, APOB, B3GA4, LDLR and PCSK9 were tested for associations with the eight metabolic QTs. The genes APOC1, APOC2, APOE, B4GA4 contain no variants with MAF<1%, and SORT1 contains only 1 rare variant site. Gene-based association analysis was not performed for these five genes. Among the 256 individuals, 33 were taking blood pressure (BP) lowering medications; and their BP levels were adjusted by adding 10 mm Hg to their SysBP and 5 mm Hg to DiasBP levels [29]. Following the same strategy as the initial LDL analysis [1], residuals for each trait were obtained and quantile-normalized after adjusting for age, age×age and sex in the entire SardiNIA cohort. The normalized residuals of the 256 samples were analyzed for associations with the four genes, i.e. APOB, B3GA4, LDLR and PCSK9. The five rare variant association tests incorporated in STAR were used to analyze the data. In addition to the secondary traits, the associations with the primary trait (i.e. LDL levels) were also analyzed. For the rare variant tests that use fixed MAF thresholds (i.e. CMC, WSS, KBAC and SKAT), variants with MAF<1% were analyzed. For VT test, variants with MAF<5% were used in the analysis. The secondary traits were also analyzed using standard linear regression that ignores the ascertainment mechanism, as a comparison to the analysis using the STAR method.
Results
Evaluation of Type I Error
Type I error for STAR was investigated when 1.) the gene region is neither associated with the primary trait nor the secondary trait. 2.) the gene region is associated only with the primary trait but not the secondary trait. The quantile-quantile plots of empirical p-values and their theoretical expectations are displayed for different rare variant tests. It can be seen that all tests incorporated in the STAR method have well controlled type I error. The p-values for the five tests are slightly conservative even when permutation is used to evaluate significance. This can occur when either the aggregate variant frequencies are low or the sample size is not sufficiently large. For example, when the primary trait effect is and residual correlation is , the type I errors for CMC, WSS, KBAC, VT and SKAT are respectively 0.048, 0.046, 0.042, 0.045 and 0.047 (Figure 1).
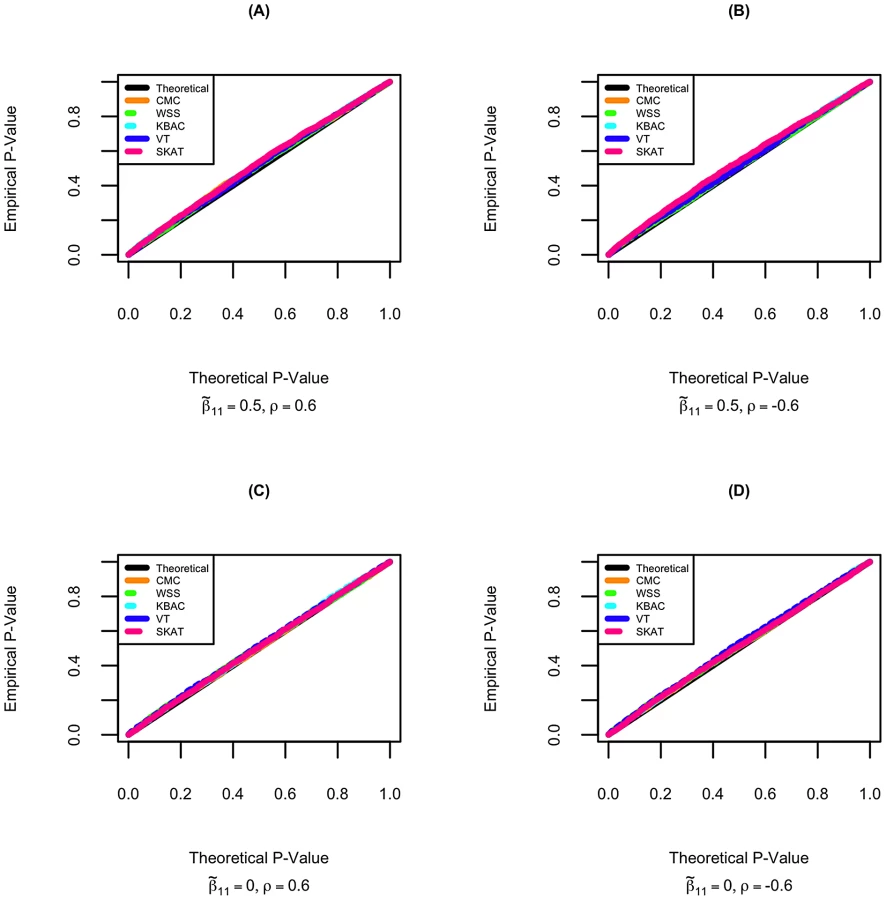
As a comparison, we also evaluated type I errors of linear regression analysis that ignores sample ascertainment mechanisms. When the gene region is not associated with the primary trait, type I errors for all rare variant tests are well controlled. However, if the gene region is also associated with the primary trait, the distribution of p-values under the null hypothesis is highly skewed and the type I errors for all tests are seriously inflated (Figure S3), which is concordant with our theoretical expectations.
Evaluation of Power
The power of detecting secondary trait associations was compared for a variety of rare variant tests (Figure 2; Figure 3; and Figures S4, S5, S6, S7). Compared to the CMC method, the extended SKAT, WSS, KBAC and VT methods in STAR can be more powerful under a broad variety of models. For example, when the primary trait is associated with the gene region with effect and trait residual correlation is , if causal variants have fixed unidirectional secondary trait effect, i.e. , the power for WSS, KBAC and VT tests are respectively 73.5%, 74.1% and 78.1%, which all have greater power than the CMC (71.5%) (Figure 2). If the secondary trait effects are bidirectional, the power for the VT (50.6%) and SKAT (54.3%) tests are much higher than that of the CMC (41.4%), and the power for KBAC (44.1%) is also slightly greater than the power for the CMC (Figure 3).
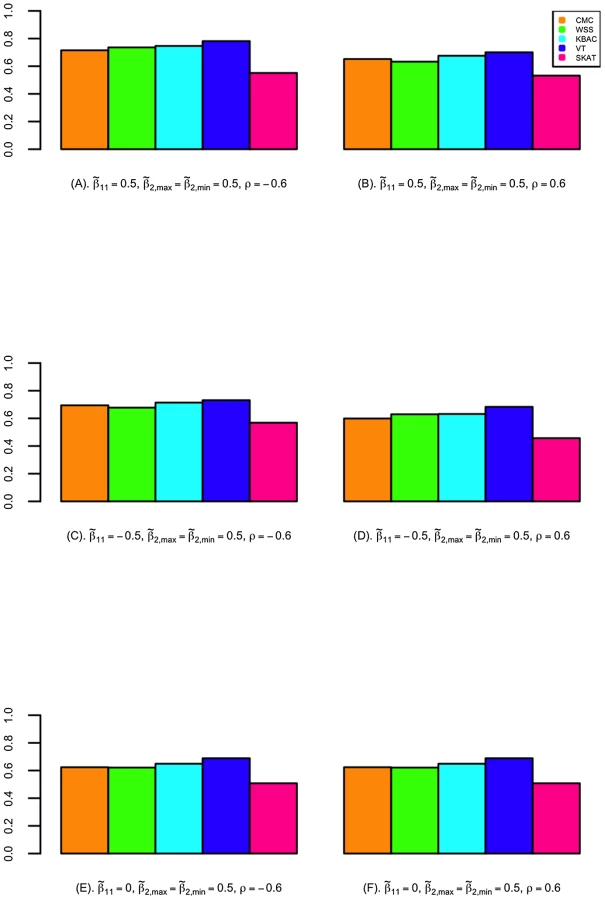
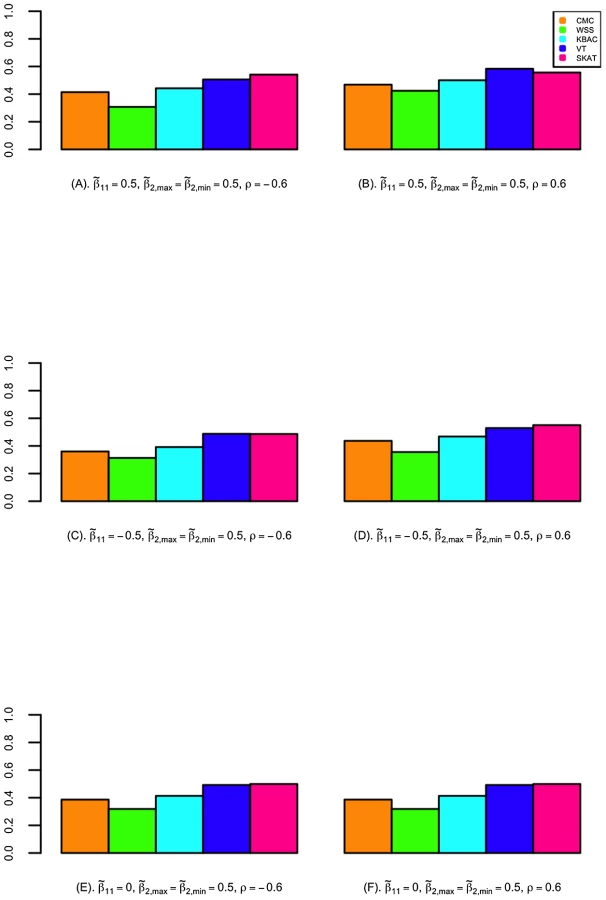
VT can be more powerful than methods that use fixed variant frequency threshold, when the secondary trait effects are unidirectional. This is because using a fixed variant frequency threshold may result in the inclusion of higher frequency non-causal variants or the exclusion of more frequent causal variants from the analyses. For example, when the primary trait effect is and trait residual correlation is , if the secondary trait effects are unidirectional and fixed with , the power for VT is 78.1%, which is considerably higher than the power for the CMC (71.5%) (Figure 2). However, the difference in power between the best performing methods is small. For instance, when the primary trait effect is and trait residual correlation is , if the secondary trait effects are unidirectional and variable with and , the power for VT is 85.9%, which is only 0.3% and 2.6% higher than the power for the WSS and KBAC.
The variance component score test SKAT is less powerful than burden tests when causal variant effects are unidirectional. For example, when , , and the causal variant effects are unidirectional with , the power for SKAT is 53.1%, which is 24.3% lower than VT and 21.6% lower than KBAC (Figure 2). However, when the causal variant secondary trait effects are bidirectional, SKAT is among the most powerful methods. For instance, if the magnitudes of the causal variant effects are inversely correlated with MAFs, when , and , the power for SKAT is 63.2%, which is much greater than the power for CMC (49.2%), WSS (51.0%), and KBAC (54.4%) and slightly higher than the power for VT (60.3%) (Figure S5).
When the gene region is associated with both the primary and secondary traits, the power to detect secondary trait associations can be greater than when the gene region is only associated with the secondary trait. This is because variants with pleiotropic effects can be more enriched through extreme sampling. For example, when secondary trait effects are , and residual correlation is , if the gene region is not associated with the primary trait, the power for CMC, WSS, KBAC, VT and SKAT are respectively 61.9%, 61.4%, 64.7%, 67.7% and 49.7% (Figure 2). However, if the gene region is also associated with primary trait with effect , the power for the five tests increases to 65.3%, 63.3%, 67.7%, 70.1% and 53.1% respectively (Figure 2). Therefore, the power for detecting secondary trait associations can also be increased through sequencing samples with extreme primary trait values.
Application of STAR to Meta-Analysis
The power and type I errors for STAR were evaluated for a simulated meta-analysis of three studies. As shown in (Figure S8), the empirical p-values and their theoretical expectations are well aligned on the quantile-quantile plot. Under a significance level of α = 0.05, the type I errors for the five rare variant tests are CMC (0.051), WSS (0.049), KBAC (0.049), VT (0.047), SKAT (0.051), which are well controlled. Due to the small sample size that is used, the type I errors for analyzing each individual study can still be slightly conservative. For example, in study 1, where causal variant effect for the primary trait is −0.5 and the correlation between the primary and secondary traits is 0.6, the type I errors for the five tests are respectively: CMC (0.046) WSS (0.044), KBAC (0.046), VT (0.047) and SKAT( 0.045).
We also evaluated the power of the STAR method under the alternative hypothesis (Figure S9). It can be seen that the power for meta-analysis is always higher than the power for each individual study, which highlights the benefit of combining multiple studies to detect associations with commonly measured traits.
Analyses of SardiNIA Sequence Dataset
Sequence data from the SardiNIA project were analyzed to detect associations with multiple lipids and metabolic traits. First, association analyses were carried-out for the primary trait LDL levels (Table 1). In the original article by Sanna et al [1], extreme LDL values were dichotomized and association analyses were performed by comparing variant carrier frequencies between individuals sampled from opposite ends of the trait distribution. Only APOB was found to be nominally significantly associated with LDL (p-value 0.03). When QT values are analyzed instead of the dichotomized trait and more powerful association methods are used, the power to detect associations with the primary trait can be increased. For the association with APOB, the p-values for the five tests are , , , , , . Additionally a significant association with LDLR that was not previously detected was also observed (, , , , ). Among the tests that were used to analyze the association between LDLR and LDL, VT gives the smallest p-value. On the other hand, for the association with APOB, the score statistics from VT are maximized at the same MAF threshold as used by the other tests (i.e. 1%). In this case, the CMC test gives the most significant p-value.
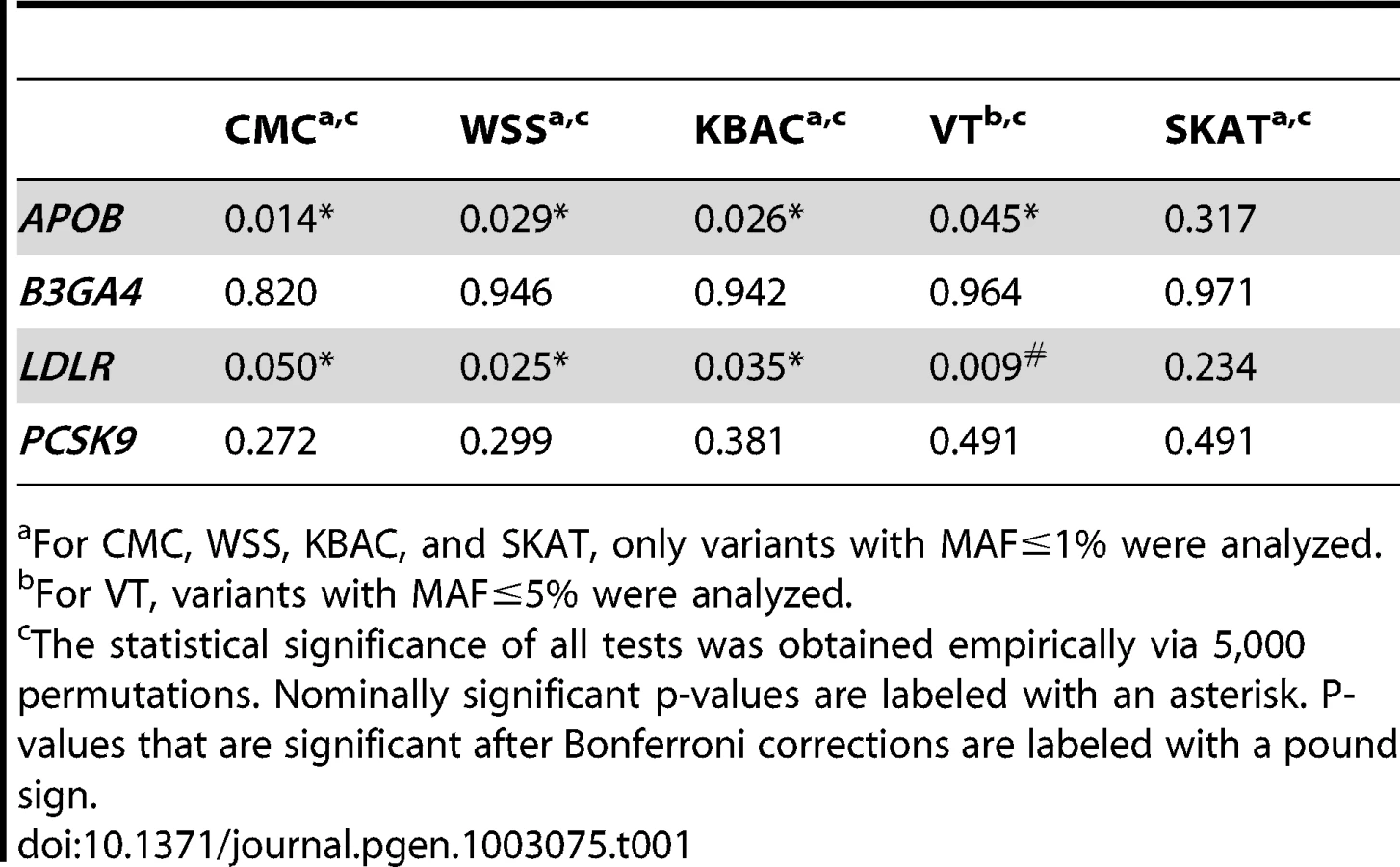
Next we analyzed secondary trait associations with the four genes, i.e. APOB, B3GA4, LDLR and PCSK9 (Table 2). One significant association, i.e. the association between LDLR and SysBP, is identified by CMC, WSS and KBAC after applying a Bonferroni correction for testing multiple genes and traits. The p-values for VT and SKAT are also nominally significant (, , , , ). The score statistics in VT are maximized at the MAF cutoff 1%. In this scenario, the p-value of VT is not as significant as that of CMC, WSS and KBAC, because the burden test statistics are not increased at alternative frequency thresholds and a penalty for multiple testing is paid.
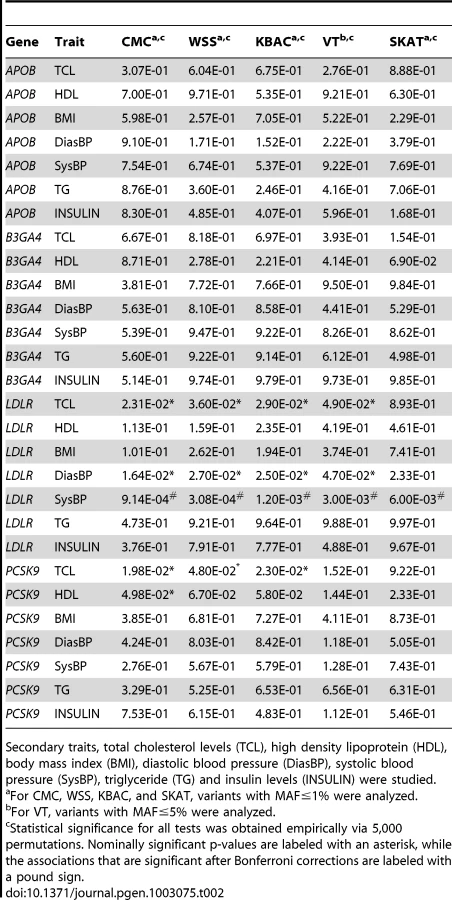
It is interesting to note that LDLR is associated with both the primary trait LDL and the secondary trait SysBP, which are correlated with a coefficient of 0.145 (Table S1). It is possible that a portion of the rare variants in LDLR have pleiotropic effects and are enriched in the dataset via selective sampling on the primary trait, which increase the power for detecting secondary trait associations.
We also compared the analysis using STAR and standard linear regressions (Table S2). Due to the small sample sizes that are used, we did not observe an excess of false positive signals for the naïve linear regression analysis. However, we noted that for the association between LDLR and SysBP, the p-values from STAR are smaller. In addition, for the associations between LDLR, PCSK9 and TCL that were previously implicated in genome-wide association studies [30], the p-values from STAR are also more significant.
Discussion
In this article, we present a likelihood model which can be used to analyze secondary trait associations in selected samples. The method corrects for the bias in the distribution of the secondary traits induced by selective sampling. All rare variant association analysis methods can be extended within the STAR framework. STAR makes it possible to apply more powerful rare variant association tests for the analysis of secondary trait and allows jointly analyzing cohorts that were ascertained for different primary traits. The power for detecting associations with secondary traits can be greatly enhanced. In addition to performing gene-based association analysis, the STAR method and STARSEQ software can also be applied to detect single variant associations (data not shown).
Currently, many sequence based genetic studies are being performed to detect associations with complex traits. Due to the high cost of sequencing, the sample sizes for many of these studies are small. It was previously shown that in order to have sufficient power (e.g. >80%) to detect association with rare variants in an exome-wide study, in some cases it is necessary to sequence at least 10,000 samples with extreme traits from a cohort of 100,000 [25]. However, both the cohort size and the cost of sequencing exceed the capacity of most studies. Therefore to increase power it is important that multiple studies can be jointly analyzed. The STAR method is particularly useful, since cohorts that are ascertained for diverse primary traits using different study designs can be jointly analyzed.
Previously CMC and GRANVIL tests were extended for analyzing secondary traits with p-value being evaluated analytically [21]. Many of the rare variant association methods implemented in STAR can be more powerful than CMC and GRANVIL. In fact, despite being computationally efficient, CMC and GRANVIL can be underpowered when a large portion of the variants in the gene region are non-causal or when the genetic effects of causal variants are bidirectional. Other methods such as SKAT may perform better in these scenarios. In addition, through assigning weights to different variant sites, variants that are potentially causal can be assigned higher weights, which can help to distinguish causal from non-causal variants. When variable selection based methods are used, the set of variants where the Z-score statistics are maximized can be selected and tested. These methods can be more robust against the inclusion of non-causal variant in the analysis, and also potentially be more powerful than CMC and GRANVIL methods even after adjusting for multiple comparisons.
Permutation algorithm is often a necessary ingredient for rare variant association tests. Even if asymptotic approximations exist for some rare variant association tests such as SKAT and CMC, they may not be accurate and type I errors may be inflated or deflated [23]. This is because the asymptotic distribution for the test statistic can be affected by the number of rare variant sites and variant site frequency spectrums [17], [23]. In practice, there can be considerable variation in the number of variant sites and frequencies within a gene region [31]. It is possible that an asymptotically valid test has inflated or deflated type I errors when genetic regions with only a few variant alleles are analyzed. Therefore, when a significant analytical p-value is obtained, it is necessary to empirically confirm the result using permutation.
Under the STAR framework, we compared the power of several rare variant tests for analyzing secondary traits in selected samples. It is clear from our comparisons that when causal variants have unidirectional effects, burden tests perform better than SKAT. However, when variants with effects in opposite directions are present, SKAT can be more powerful than burden test based methods. Given that the goal of the article is to introduce a method for analyzing secondary traits in selected samples, rather than to compare different rare variant tests, our simulations are not as comprehensive as some existing reviews, such as Basu and Pan [32] and Ladouceur et al [33]. However, based upon the simulation scenarios that we considered, it is clear that the power for detecting associations can be greatly improved through the STAR model. In addition, our conclusions for comparing multiple rare variant tests are also compatible with the comprehensive reviews, in that there is not a consistently most powerful rare variant test and the difference in power between top performing methods is usually small. In addition to the simulation experiments, it is also important to examine and compare the performance of different methods in large scale sequencing studies, such as the NHLBI-ESP.
In the analysis of the SardiNIA dataset, we adjusted the blood pressure for individuals undergoing antihypertensive therapy. The rank of sample blood pressure traits was only slightly changed after the adjustment. Given that we quantile-normalized the trait prior to the association analysis, the impact of the adjustment on the result is very minimal. In order to evaluate the robustness of the results, we also analyzed the associations with blood pressure when no adjustments were made, and the results are very similar (data not shown). A significant association was identified between rare variants in LDLR and secondary trait SysBP, where carriers of rare variants in the LDLR gene tend to have lower SysBP levels. In fact, the LDLR gene has also been shown to be strongly associated with reductions of SysBP among the patients that receive atenolol, an antihypertensive drug [26]. These discoveries imply that variants in the LDLR gene may influence the etiology of SysBP. LDLR is potentially an important gene target for blood pressure treatment. In order to replicate signals [22] that were found in the SardiNIA cohort, many current large scale sequencing studies can be considered, such as the NHLBI-ESP etc. In addition to replicating associations, there is also great scientific interest in exploring whether rare causal variants identified in a founder population are identical to those from out-bred populations [34].
With the large scale application of next generation sequencing to study complex traits, samples from many existing cohorts will be sequenced. There can be insufficient power for analyzing associations in each individual study. It would be highly beneficial if samples from multiple cohorts can be combined for analyzing commonly measured traits. STAR is thus important and will greatly accelerate the process of identifying genes involved in complex trait etiology.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. SannaS, LiB, MulasA, SidoreC, KangHM, et al. (2011) Fine mapping of five loci associated with low-density lipoprotein cholesterol detects variants that double the explained heritability. PLoS Genet 7: e1002198 doi:10.1371/journal.pgen.1002198.
2. RaychaudhuriS, IartchoukO, ChinK, TanPL, TaiAK, et al. (2011) A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat Genet 43: 1232–1236.
3. RivasMA, BeaudoinM, GardetA, StevensC, SharmaY, et al. (2011) Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet 43: 1066–1073.
4. JiW, FooJN, O'RoakBJ, ZhaoH, LarsonMG, et al. (2008) Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet 40: 592–599.
5. HarismendyO, BansalV, BhatiaG, NakanoM, ScottM, et al. (2011) Population sequencing of two endocannabinoid metabolic genes identifies rare and common regulatory variants associated with extreme obesity and metabolite level. Genome Biol 11: R118.
6. DarvasiA (2006) Closing in on complex traits. Nat Genet 38: 861–862.
7. PlominR, HaworthCM, DavisOS (2009) Common disorders are quantitative traits. Nat Rev Genet 10: 872–878.
8. CauchiS, NeadKT, ChoquetH, HorberF, PotocznaN, et al. (2008) The genetic susceptibility to type 2 diabetes may be modulated by obesity status: implications for association studies. BMC Med Genet 9: 45.
9. IoannidisJP, ThomasG, DalyMJ (2009) Validating, augmenting and refining genome-wide association signals. Nat Rev Genet 10: 318–329.
10. McCarthyMI, AbecasisGR, CardonLR, GoldsteinDB, LittleJ, et al. (2008) Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 9: 356–369.
11. Bouatia-NajiN, RocheleauG, Van LommelL, LemaireK, SchuitF, et al. (2008) A polymorphism within the G6PC2 gene is associated with fasting plasma glucose levels. Science 320: 1085–1088.
12. ElliottP, ChambersJC, ZhangW, ClarkeR, HopewellJC, et al. (2009) Genetic Loci associated with C-reactive protein levels and risk of coronary heart disease. JAMA 302: 37–48.
13. HanF, PanW (2010) A data-adaptive sum test for disease association with multiple common or rare variants. Hum Hered 70: 42–54.
14. MadsenBE, BrowningSR (2009) A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet 5: e1000384 doi:10.1371/journal.pgen.1000384.
15. LiuDJ, LealSM (2010) A novel adaptive method for the analysis of next-generation sequencing data to detect complex trait associations with rare variants due to gene main effects and interactions. PLoS Genet 6: e1001156 doi:10.1371/journal.pgen.1001156.
16. LiB, LealSM (2008) Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet 83: 311–321.
17. WuMC, LeeS, CaiT, LiY, BoehnkeM, et al. (2011) Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet 89: 82–93.
18. MorrisAP, ZegginiE (2009) An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet Epidemiol
19. BhatiaG, BansalV, HarismendyO, SchorkNJ, TopolEJ, et al. (2010) A covering method for detecting genetic associations between rare variants and common phenotypes. PLoS Comput Biol 6: e1000954 doi:10.1371/journal.pcbi.1000954.
20. LinDY, ZengD (2009) Proper analysis of secondary phenotype data in case-control association studies. Genet Epidemiol 33: 256–265.
21. LiuDJ, LealSM (2011) A flexible likelihood framework for detecting associations with secondary phenotypes in genetic studies using selected samples: application to sequence data. Eur J Hum Genet
22. LiuDJ, LealSM (2010) Replication Strategies for Rare Variant Complex Trait Association Studies via Next-Generation Sequencing. Am J Hum Genet 87: 790–801.
23. LinDY, TangZZ (2011) A general framework for detecting disease associations with rare variants in sequencing studies. Am J Hum Genet 89: 354–367.
24. PriceAL, KryukovGV, de BakkerPI, PurcellSM, StaplesJ, et al. (2010) Pooled association tests for rare variants in exon-resequencing studies. Am J Hum Genet 86: 832–838.
25. KryukovGV, ShpuntA, StamatoyannopoulosJA, SunyaevSR (2009) Power of deep, all-exon resequencing for discovery of human trait genes. Proc Natl Acad Sci U S A 106: 3871–3876.
26. LiljedahlU, LindL, KurlandL, BerglundL, KahanT, et al. (2004) Single nucleotide polymorphisms in the apolipoprotein B and low density lipoprotein receptor genes affect response to antihypertensive treatment. BMC Cardiovasc Disord 4: 16.
27. AdamsAM, HudsonRR (2004) Maximum-likelihood estimation of demographic parameters using the frequency spectrum of unlinked single-nucleotide polymorphisms. Genetics 168: 1699–1712.
28. MunafoMR, FlintJ (2004) Meta-analysis of genetic association studies. Trends Genet 20: 439–444.
29. LevyD, EhretGB, RiceK, VerwoertGC, LaunerLJ, et al. (2009) Genome-wide association study of blood pressure and hypertension. Nat Genet 41: 677–687.
30. TeslovichTM, MusunuruK, SmithAV, EdmondsonAC, StylianouIM, et al. (2010) Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466: 707–713.
31. WolfeKH, SharpPM, LiWH (1989) Mutation rates differ among regions of the mammalian genome. Nature 337: 283–285.
32. BasuS, PanW (2011) Comparison of statistical tests for disease association with rare variants. Genet Epidemiol 35: 606–619.
33. LadouceurM, DastaniZ, AulchenkoYS, GreenwoodCM, RichardsJB (2012) The empirical power of rare variant association methods: results from sanger sequencing in 1,998 individuals. PLoS Genet 8: e1002496 doi:10.1371/journal.pgen.1002496.
34. NewmanDL, HoffjanS, BourgainC, AbneyM, NicolaeRI, et al. (2004) Are common disease susceptibility alleles the same in outbred and founder populations? Eur J Hum Genet 12: 584–590.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 11
Nejčtenější v tomto čísle
- Mechanisms Employed by to Prevent Ribonucleotide Incorporation into Genomic DNA by Pol V
- Inference of Population Splits and Mixtures from Genome-Wide Allele Frequency Data
- Zcchc11 Uridylates Mature miRNAs to Enhance Neonatal IGF-1 Expression, Growth, and Survival
- Histone Methyltransferases MES-4 and MET-1 Promote Meiotic Checkpoint Activation in