
Regulatory Cross-Talk Links Chromosome II Replication and Segregation
There is little knowledge of factors and mechanisms for coordinating bacterial chromosome replication and segregation. Previous studies have revealed that genes (and their products) that surround the origin of replication (oriCII) of Vibrio cholerae chromosome II (chrII) are critical for controlling the replication and segregation of this chromosome. rctB, which flanks one side of oriCII, encodes a protein that initiates chrII replication; rctA, which flanks the other side of oriCII, inhibits rctB activity. The chrII parAB2 operon, which is essential for chrII partitioning, is located immediately downstream of rctA. Here, we explored how rctA exerts negative control over chrII replication. Our observations suggest that RctB has at least two DNA binding domains—one for binding to oriCII and initiating replication and the other for binding to rctA and thereby inhibiting RctB's ability to initiate replication. Notably, the inhibitory effect of rctA could be alleviated by binding of ParB2 to a centromere-like parS site within rctA. Furthermore, by binding to rctA, ParB2 and RctB inversely regulate expression of the parAB2 genes. Together, our findings suggest that fluctuations in binding of the partitioning protein ParB2 and the chrII initiator RctB to rctA underlie a regulatory network controlling both oriCII firing and the production of the essential chrII partitioning proteins. Thus, by binding both RctB and ParB2, rctA serves as a nexus for regulatory cross-talk coordinating chrII replication and segregation.
Published in the journal:
. PLoS Genet 7(7): e32767. doi:10.1371/journal.pgen.1002189
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002189
Summary
There is little knowledge of factors and mechanisms for coordinating bacterial chromosome replication and segregation. Previous studies have revealed that genes (and their products) that surround the origin of replication (oriCII) of Vibrio cholerae chromosome II (chrII) are critical for controlling the replication and segregation of this chromosome. rctB, which flanks one side of oriCII, encodes a protein that initiates chrII replication; rctA, which flanks the other side of oriCII, inhibits rctB activity. The chrII parAB2 operon, which is essential for chrII partitioning, is located immediately downstream of rctA. Here, we explored how rctA exerts negative control over chrII replication. Our observations suggest that RctB has at least two DNA binding domains—one for binding to oriCII and initiating replication and the other for binding to rctA and thereby inhibiting RctB's ability to initiate replication. Notably, the inhibitory effect of rctA could be alleviated by binding of ParB2 to a centromere-like parS site within rctA. Furthermore, by binding to rctA, ParB2 and RctB inversely regulate expression of the parAB2 genes. Together, our findings suggest that fluctuations in binding of the partitioning protein ParB2 and the chrII initiator RctB to rctA underlie a regulatory network controlling both oriCII firing and the production of the essential chrII partitioning proteins. Thus, by binding both RctB and ParB2, rctA serves as a nexus for regulatory cross-talk coordinating chrII replication and segregation.
Introduction
Efficient linkage of chromosome replication and chromosome segregation is necessary for all dividing cells. It is particularly important for maintaining balanced genetic content in organisms with more than a single chromosome, which includes a number of bacterial orders (e.g., Vibrionaceae, Photobacteriaceae [1]). However, there is relatively little knowledge of factors and mechanisms that link replication and segregation of bacterial chromosomes. For the gram-negative enteric pathogen Vibrio cholerae, whose genome is comprised of two circular chromosomes [2], distinct mechanisms that control the replication and segregation of each chromosome have been described, but no mechanisms for linking or coordinating these processes have been identified.
The two V. cholerae chromosomes have distinct initiator proteins that are specific for their target chromosomes. The initiator of chromosome I (chrI) replication is DnaA, a conserved AAA+ ATPase protein found in nearly all eubacteria [3]–[6]. V. cholerae DnaA binds and melts the origin of replication of chrI (oriCI) but not that of oriCII, the origin of replication of chromosome II (chrII) [7]. It is likely that regulation of DnaA-mediated initiation of V. cholerae chrI parallels DnaA-dependent control of replication initiation in Escherichia coli [6], [7].
The initiator of chrII replication is RctB, a protein that is encoded near oriCII and conserved among, but restricted to, the Vibrionaceae/Photobacteriaceae (Figure 1A) [3]. RctB specifically binds and opens oriCII DNA in vitro, and its overexpression in V. cholerae leads to overinitiation of chrII but not chrI [4], [7]. RctB can bind and hydrolyze ATP, despite a lack of known ATP binding motifs; however, unlike other ATPase initiator proteins, the ATP-bound form of RctB is inactive for oriCII replication [7]. RctB activity is also negatively regulated by rctA, a neighboring gene [8]. Although rctA is transcribed [3] and was originally annotated as an ORF [2], it does not seem to encode a functional protein; instead at least one role of rctA appears to be as a DNA site for binding RctB, perhaps thereby titrating the initiator from oriCII [8]. Overall, the regulation of RctB activity and chrII replication initiation, which can be modulated by the factors noted above, by transcription within the oriCII region [8], and by additional proteins such as Dam and SeqA [3], [9], is complex and incompletely understood.

Although distinct proteins govern initiation of chrI and chrII replication, the replication of the two V. cholerae chromosomes is thought to be coordinated with the cell cycle, which should facilitate maintenance of genomic balance [10]–[12]. Genomic integrity is also promoted by chromosome-specific par systems, which have been implicated in the subcellular localization and/or partitioning of the respective oriC regions of each chromosome [13]–[16]. These systems consist of ParA ATPases, DNA-binding ParB proteins, and cis-acting ParB binding sites, parS ([17], [18] for review). The two V. cholerae ParB proteins (ParB1 and ParB2, encoded on chrI and chrII, respectively) recognize distinct parS sequences (parS1 and parS2, respectively) [15]. While the nucleotide sequence of parS1 is identical to the ‘universal’ parS sequence originally described in Bacillus subtilis [19], the nucleotide sequence of parS2 is restricted to vibrio and photobacteria species [15], [20]. All but one of V. cholerae's 10 consensus parS2 sites lie within chrII, and most of them are located proximal to oriCII. Interestingly, one of the parS2 sites, designated parS2-B, is located within rctA, suggesting the possibility that this site could provide a basis for coordination of the control of chrII replication and segregation. Individual parS2 sites are not essential for V. cholerae viability ([15] and data not shown); however, deletion of the chrII parAB2 locus results in loss of chrII and cell death [16].
Here we explore how RctB interacts with rctA and how rctA negatively regulates chrII replication. Our observations suggest that RctB has at least two DNA binding domains - one for binding to oriCII and the other for binding to rctA. RctB lacking its C-terminus fails to bind rctA in vitro and its replicative activity is not inhibited by rctA in vivo. Notably, the inhibitory effect of rctA on RctB could also be alleviated by binding of ParB2 to the parS2 site within rctA. Furthermore, ParB2 and RctB binding to rctA inversely alter the expression of the parAB2 genes. Together, our findings suggest that fluctuations in binding of the partitioning protein ParB2 and the chrII initiator RctB to rctA underlie a regulatory network controlling both oriCII firing as well the production of the essential partitioning proteins ParA2 and ParB2. Thus, by binding both RctB and ParB2, rctA serves as a nexus for regulatory cross–talk coordinating chrII replication and segregation.
Results
A screen for factors enabling replication of an oriCII-based plasmid containing rctA
Previous studies have established that plasmids harboring oriCII (defined as the region between rctA and rctB ([3]; see Figure 1) as the sole origin of replication can replicate in E. coli as long as RctB is present, and that such replication can be inhibited by the presence of rctA either in cis or in trans [8], [21]. We utilized a similar approach to further dissect the molecular basis of rctA's inhibition of oriCII-based replication. The replication capacity of plasmids that included rctB, oriCII, and various additional linked sequences were assessed by their efficiency of transformation into heterologous (E. coli) host strains containing either a control vector or one that overexpressed RctB (Figure 1B). Using this system, we obtained transformants within 24 hrs of introducing a plasmid that lacked rctA (pYB289), regardless of whether RctB was overexpressed (Figure 1B[a]). In contrast, no transformants were detectable 24 hrs after introduction of a plasmid that contained rctA (pYB292) unless the RctB expression construct was also present (Figure 1B[b]), consistent with the suggestion that sequestration of RctB by rctA reduces its replicative activity [8].
Notably, after ∼48 hrs, rare transformants were obtained with pYB292 even in the absence of RctB overexpression. Most of these colonies could be re-streaked, and plasmid DNA was recovered and sequenced from sixty five transformants. Sixty of these plasmids carried mutations that fell into one of three groups: 1) deletions of rctA (n = 6), 2) substitutions in the rctB sequence that result in amino acid substitutions in RctB (n = 34), and 3) substitutions or deletions in rctB that result in truncations of the carboxyl terminus of RctB (n = 20) (Figure 2 and Table S1). In general, strains carrying these plasmids grew at wild-type rates following the initial 24 hr lag in their detection, suggesting that the mutations within RctB did not impair its replicative capacity. Notably, none of the mutations mapped to the oriCII sequence per se, an observation that is consistent with the idea that an rctA transcript or protein does not act in trans on oriCII. In the remaining 5 cases, mutations were likely present in the host E. coli chromosome, since the purified oriCII plasmids (which did not harbor mutations) could be re-transformed into the DH5α strain they were isolated from (after plasmid curing) but not into a fresh isolate of DH5α.

The C-terminus of RctB interacts with rctA
The prevalence (20 of 60 clones) of RctB truncations among pYB292 derivatives whose replication did not require RctB overexpression (Figure 2, Table S1) suggested that the C-terminal part of RctB might be required for its interaction with, and inactivation by, rctA. However, the normal replication of plasmids containing such truncations (predicted to remove at least 41, and at most 159, amino acids from the C-terminus of RctB) indicates that truncated RctB retains the capacity to melt oriCII and initiate chrII replication. Together, these observations raise the possibility that RctB has multiple sites and/or modes for interacting with DNA. To explore this hypothesis, we compared the binding of His-tagged full length RctB and RctB(Δ500–658), which lacks 159 amino acids of the protein's C-terminus (hereafter referred to as RctB[ΔC159]), to oriCII and rctA, using an electrophoretic mobility shift assay (EMSA). Both proteins readily bound to oriCII, and they appear to have a similar affinity for this sequence, although RctB[ΔC159] appears to have a greater tendency to form multimeric complexes on the DNA (Figure 3A). In contrast, while wild type RctB bound to the rctA probe, almost no binding of RctB[ΔC159] was detected (Figure 3A). Together, these observations suggest that RctB has at least two DNA binding domains; one, which binds oriCII, is contained within RctB[1–499] and can mediate oriCII-based replication, while the other, which binds rctA, is at least partially contained within, or dependent upon, sequences within RctB[500–658]. We were unable to demonstrate binding of RctB[500–658] to rctA, suggesting that additional regions of RctB likely also contribute to rctA binding. Thus, although some sequence similarity has been noted between potential RctB target sites within oriCII and rctA [8], our data raises the possibility that RctB actually recognizes two distinct sequences. Additionally, our data provides genetic and biochemical support for the hypothesis that RctB binding to rctA is the basis for rctA's negative influence on oriCII-based replication.

ParB2 binding to parS2-B alleviates the negative influence of rctA on oriCII replication
Similar to other chromosomal parS sites, most parS2 sites are located proximal to oriCII on chrII [15], [21]. One of these (designated parS2-B) is found within the originally annotated rctA sequence (Figure 1A, ref. [15]). A parS2 site is present at a similar position relative to oriCII in the genomes of multiple other vibrio species [15], despite an overall lack of conservation of the surrounding sequence. We hypothesized that ParB2 binding to this parS2 site might influence binding of RctB to rctA, and perhaps thereby regulate oriCII-based replication. This possibility was investigated by measuring the effect of ParB2 on the efficiency with which various oriCII-related replicons could be transformed into E. coli. Overexpression of ParB2 had a minimal effect on the transformation of pYB289, consistent with the absence of rctA/parS2B within this construct (Figure 1B[a]). However, overexpression of ParB2 caused a dramatic increase in the efficiency with which the rctA-containing plasmid pYB292 could be introduced into E. coli (Figure 1B[b]). The effect of ParB2 expression was abolished when an alternate plasmid, pYB558, in which the parS2-B site was mutated to parS2X [15], was transformed instead (Figure 1B[c]). Transformants were still obtained with pYB558 when it was introduced into a strain that overexpressed RctB but not when it was introduced into a strain containing an empty vector, suggesting that the mutation in parS2-B did not interfere with binding of RctB, and thus that the two proteins do not recognize identical sequences (Figure 1B[b]). Data from the transformation assay was consistent with results from EMSAs, which revealed that ParB2 bound with high affinity to wild type rctA but not rctA containing parS2X, while RctB bound to both probes (Figure 3A). Overall, these data indicate that ParB2 binding to parS2-B can mask the negative effect of rctA upon replication of oriCII-based replicons.
RctB and ParB2 can simultaneously bind rctA
The simplest explanation for increased transformation efficiency of pYB292 in the presence of overexpressed ParB2 is that binding of ParB2 to rctA interferes with binding of RctB to this site, and thereby makes more RctB available for replication initiation at oriCII. However, EMSA analyses did not provide direct support for this hypothesis. Instead, they indicate that RctB and ParB2 can bind simultaneously to rctA (Figure 3B and Figure S1). DNase I protection experiments confirmed that RctB can bind to rctA, but a specific region of binding was not observed (Figure 4). Instead, when ∼40–80 ng of RctB were added to the assay, several non-adjacent nucleotides that were distributed irregularly throughout the rctA sequence were protected from DNase I digestion (Figure 4, arrowheads). When higher amounts of RctB (∼160–640 ng) were added, the protection of individual bands became less pronounced and much of the fragment exhibited a degree of protection, including the parS2-B site. In contrast, ParB2 protected a ∼20 bp continuous stretch of DNA around the parS2-B site (Figure 4). Inclusion of both RctB and ParB2 in the DNAse I protection assays resulted in additive protection, consistent with simultaneous binding of both proteins to rctA. Additionally, DNAse I-hypersensitive sites (Figure 4, arrows) observed in the presence of ParB2 alone became protected upon inclusion of RctB in the reaction, suggesting that RctB can alter rctA structure even when ParB2 is bound. Collectively the EMSA and footprinting assays show that RctB and ParB2 can simultaneously bind to rctA. However, given the similar patterns of protection of the parS2-B region by the two proteins, it is difficult to ascertain whether ParB2 interferes with RctB's binding to this domain within rctA.

Titration of ParB2 by ectopic parS2 sites reduces oriCII-based replication
In order to assess the roles of ParB2, parS2 and rctA at more physiological levels in vivo, we generated an additional construct (pYB404) for the transformation assay that contained all 6 kb of DNA from parB2 through rctB (Figure 1A), and thereby enabled expression of ParB2 from its endogenous promoter. In contrast to pYB292, pYB404 replicated in E. coli even without overexpression of RctB or ParB2, despite the presence of rctA in this construct (Figure 5A). Thus, ParB2 produced from its own promoter appears to be sufficient to overcome the negative effect of rctA on oriCII-mediated replication. However, overexpression of either RctB or ParB2 in the E. coli strain did increase the number of transformants obtained, perhaps because limiting amounts of these proteins are present when the plasmid is becoming established (Figure 5A).
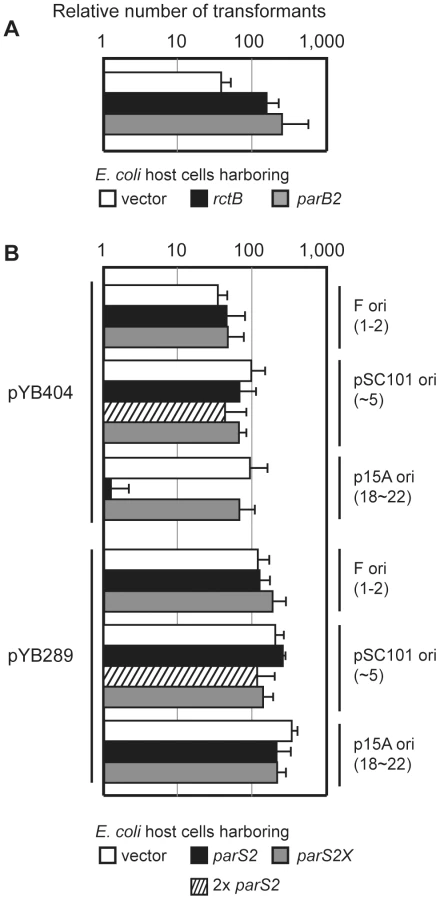
We also assessed the influence on transformation efficiency of supplying additional copies of parS2 from one of three plasmids with different origins of replication and copy numbers: F (1–2 copies/cell), pSC101 (∼5 copies/cell) and p15A (18∼22 copies/cell) [22]. The number of pYB404 transformants obtained was not altered if the ectopic parS2 sequences were in the F-plasmid or even in pSC101 harboring two copies of parS2 separated by 1.4 kb (pYB451, yielding ∼10 ectopic copies of parS2 (Figure 5B). However, there was a marked decrease in the efficiency of pYB404 electroporation when the ectopic parS2 sites were provided from the moderate-copy-number vector p15A (Figure 5B, p15A ori). In contrast, the parS2X sequences, which do not bind ParB2, did not alter pYB404 transformation efficiency when supplied from any of the vectors. (Figure 5B, gray bars). Neither wild type nor mutant parS2 sequences altered the transformation efficiency of a vector (pYB289) that lacks rctA. Thus, the presence of 10 parS2 sequences (notably, their level within the V. cholerae genome) is compatible with replication of a oriCII-based replicon containing rctA/parS2B. However, an increase to 20 copies (e.g., as should happen following chrII replication) interferes with oriCII-based replication, presumably because ParB2 is titrated away from the parS2-B site at the origin and thus can't counteract rctA-dependent repression. This finding suggests that ParB2 makes an important contribution to controlling replication as well as partitioning of V. cholerae chrII.
ParB2-parS2 and RctB-rctA interactions contribute to oriCII replication control in V. cholerae
To further explore the contributions of rctA and parS2-B to regulation of chrII replication, we constructed ΔrctA V. cholerae (YBB995) and parS2-B::parS2X V. cholerae (YBB999). These mutant strains did not have detectable growth defects compared to the wild type V. cholerae strain N16961 (Figure S2), indicating that rctA or parS2-B mediated control of oriCII is not critical for V. cholerae viability in rich media. However, quantitative PCR assays measuring the oriCII : oriCI ratio revealed that these mutations influence oriCII replication. The ΔrctA cells exhibited a higher oriCII : oriCI ratio compared to wild type cells (Table 1), in agreement with a previous report [23]. In contrast, the parS2-B::parS2X strain YBB999 had a modest but statistically significant (p = 0.001) reduction in the oriCII : oriCI ratio compared to the wild type (Table 1). Both of these results are consistent with our findings using the heterologous host and support the model that ParB2 binding to parS2-B inhibits the negative effect that rctA exerts on RctB-mediated oriCII replication.
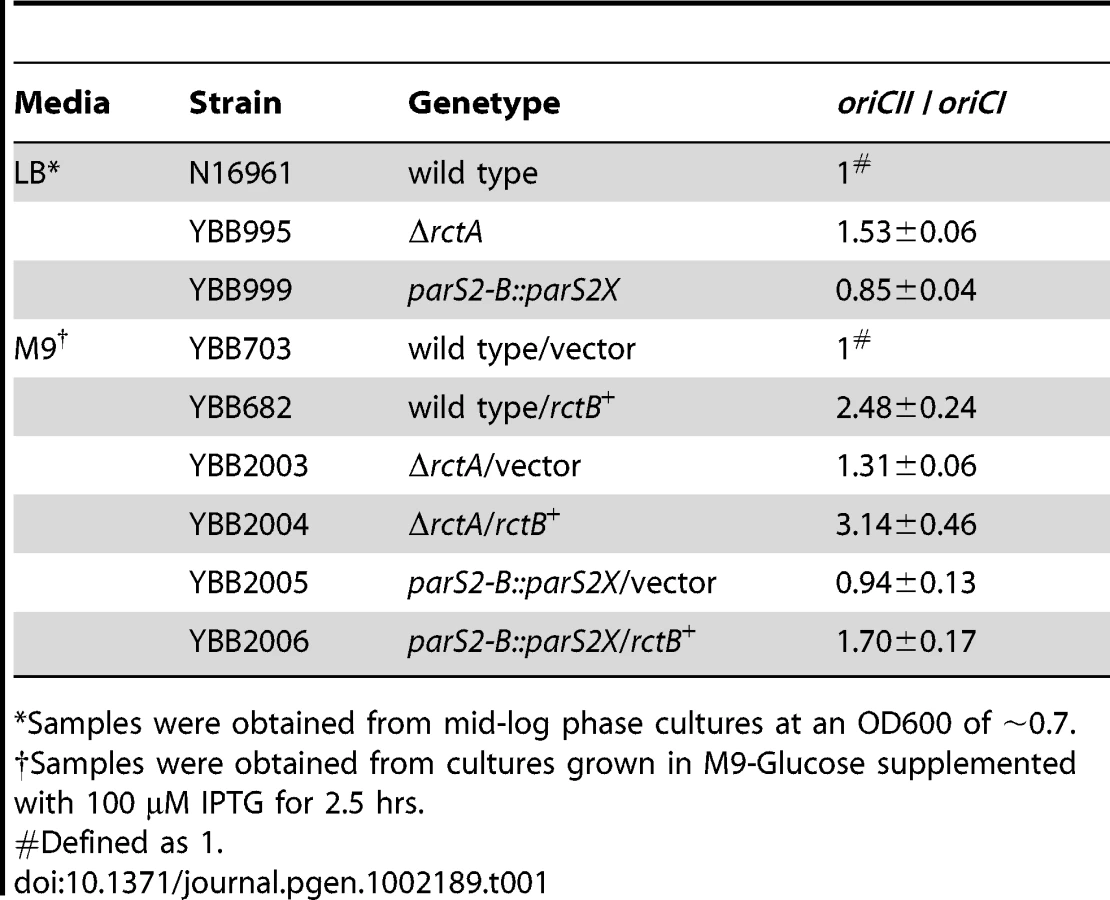
The causes and consequences of chrII overinitiation have not been thoroughly analyzed. However, previous work revealed that extreme overinitiation of oriCII mediated by the RctB mutant RctB[R269S] resulted in a block in V. cholerae cell division, manifest as cell elongation along with a marked decrease in viability [7]. In contrast, modest overinitiation of oriCII resulting from overexpression of wild type RctB had virtually no effect on cell viability or morphology (Figure 6A, 6C; see [4], [7], [23]). Similarly, the modest overinitiation of oriCII caused by the rctA deletion in YBB995 did not have a detectable affect on cell viability or morphology (Figure 6; [23]). However, deletion of rctA sensitized V. cholerae to the deleterious effects of RctB overexpression. In the ΔrctA background, RctB overexpression reduced cell viability, particularly in M9 media (Figure 6B), led to an increase in the oriCII : oriCI ratio (Table 1), and led to cell filamentation (Figure 6D). In contrast, in the parS2-B::parS2X background, RctB overexpression had little discernible influence on cell division or viability (Figure 6A, 6B), as might be expected given that the basal level of chrII replication initiation in this strain is even lower than in the wild type strain, whose viability was also unimpaired by RctB overexpression. Collectively, these data suggest that V. cholerae can adapt to some variability in RctB levels and availability, and that numerous regulatory processes are geared towards preventing the toxic effects of overinitiating replication of chromosome II.

RctB and ParB2 control transcription of parAB2
Additional forms of cross-talk between RctB and the parAB2 locus are evident from analyses of oriCII-region transcription, which revealed that binding of either ParB2 or RctB to rctA altered parAB2 promoter activity. As has been observed for several additional parAB systems [24]–[26], ParB2 significantly decreased the expression of a PparAB2 - lacZ fusion (more than 4-fold; Figure 7). This repression was abolished when parS2-B was mutated to parS2X (Figure 7), strongly suggesting that ParB2 binding to parS2-B is required for autorepression of the parAB2 locus. In contrast, RctB modestly enhanced expression of parAB2 (Figure 7, p = 0.0003), an effect that does not appear to depend on the parS2-B site in rctA. Thus, RctB binding to rctA may, despite initially limiting the amount of initiator protein available for replication initiation, ultimately promote replication, as such binding prompts expression of ParB2, which can counter repression of replication. Additionally, these results suggest that cross-talk between pathways controlling replication and partitioning is bidirectional, which is likely to enhance the coordination of these two critical processes.

Discussion
Collectively, our observations suggest that control of V. cholerae chrII replication and segregation is linked by a regulatory circuit that involves ∼6 kb of sequence (and its products) that flank oriCII and includes parAB2, rctA, and rctB. The primary agent governing replication initiation is RctB; however, initiation can also be influenced by a previously characterized partitioning protein, ParB2, which we now show counteracts rctA's inhibitory effect upon chrII replication. Analogously, the autoregulatory parAB2 locus is the primary determinant of chrII segregation; however, this process can also be influenced by RctB, which activates parAB2 expression. It appears likely that the cross-talk between these two systems both prevents extreme fluctuations in protein and chromosome abundance, and enables coordination of chromosome replication and partitioning.
Binding of the chrII replication initiator RctB to the chrII origin and surrounding sequences appears to be more complex than was previously recognized. Our analyses indicate that RctB may in fact have multiple DNA binding modes/domains, which recognize distinct sequences. RctB lacking its C-terminus (as many as 159 amino acids) retained the capacity to bind to oriCII and to initiate replication at this site. However, both EMSA experiments and DNase I protection assays (Figure S3) revealed that RctB[ΔC159] is unable to bind to rctA. Residues outside of the C-terminal 159 amino acids are also likely to contribute to rctA binding, although they remain to be identified. The presence of distinct DNA binding domains within the N - and C - terminal parts of RctB introduces the possibility that a single RctB can simultaneously bind to rctA and oriCII. Additional studies are needed to assess whether the binding of RctB to these two sites introduces a bend in the DNA between them. Studies to assess whether the point mutations within rctB that enable a bypass of rctA-mediated replication inhibition do so by altering binding to rctA or instead alter other aspects of RctB's activity or its affinity for oriCII are also warranted.
To date, precise sequences targeted by RctB have not been identified; it has been speculated that this protein recognizes some short (11 and 12-mer) repeated sequences within the origin and surrounding sequences [3], [8]. Our footprinting analyses suggest that multiple RctB proteins are interacting with the DNA; however, the repeats do not seem to be the principal target of RctB's C-terminal DNA-binding domain, as many protected sites lie outside of the repeats. The distribution of sites within rctA that are protected from DNase I digestion by RctB is unusual, in that RctB appears to interact with multiple non-continuous bases throughout this sequence. One possible explanation for this result is that RctB binding alters the secondary structure of rctA DNA. Although the DNase I protection assays suggest that multiple RctB proteins interact with rctA, especially at high protein concentrations, EMSAs only revealed a single shifted band. The different assay conditions (e.g. the presence of competitor DNA in the EMSAs) may explain this apparent discrepancy. Additional analyses are needed to assess the binding sites for RctB in oriCII.
Our studies confirmed previous reports that rctA inhibits replication of oriCII-based replicons. The inhibitory effect of rctA can be overcome by overexpression of RctB (Figure 1B; [8]). Unexpectedly, our work revealed that rctA's effect can also be mitigated by overexpression of ParB2, which recognizes a parS2 site (parS2-B) within rctA. At least three non-mutually exclusive models can explain how ParB2 abolishes rctA inhibition of replication. One possibility is that ParB2 competes with RctB in binding to rctA, resulting in more free RctB that can interact with oriCII. However, both EMSA and DNase I protection assays demonstrated that ParB2 does not block all RctB binding within rctA in vitro; at most, only the subset of RctB binding sites in parS2-B are blocked by the presence of ParB2. However, these in vitro assays may not fully reflect binding dynamics in vivo in which binding to rctA maybe influenced by the adjacent oriCII site and by additional factors such as IHF. An alternative possibility is that RctB binding to rctA alters the secondary structure of the oriCII region in a manner that inhibits replication. ParB2 binding to parS2-B could counteract RctB-mediated remodeling of oriCII, thereby promoting replication. ParB2 might also alter the extent to which rctA is transcribed, which has also been shown to influence rctA's effectiveness as a replication inhibitor [8]. It is unlikely that the effect of ParB2 upon replication is mediated by a direct interaction between this protein and RctB, as no such interaction was detected using a bacterial two hybrid system (Figure S4). Regardless of the mechanism by which it acts, it is clear that ParB2, previously described as a key agent mediating chrII segregation, also contributes to regulation of chrII replication, thereby enabling linkage of these cellular processes.
We hypothesize that the organization of this regulatory scheme is adapted to accommodate the cell cycle. As ParB2 accumulates, perhaps to amounts that are sufficient to enable chrII segregation, the repressive effects of rctA are relieved, and initiation of chrII replication ensues. Subsequently, ParB2 is re-distributed among the newly synthesized parS2 sites, and its binding to parS2-B is reduced, enabling rctA to inactivate RctB, and thereby reducing the ability of RctB to initiate replication.
Cross-talk between chrII replication and partitioning is also evident at the level of transcription. The parAB2 locus is autorepressed by parB2, as has been observed for other parAB loci [24]–[26]; in addition, we demonstrate that parAB2 transcription is activated by RctB. The contrasting effects of these two regulators are likely to rebalance ParAB levels if their abundance becomes aberrantly elevated or reduced.
Undoubtedly, additional factors and mechanisms intersect with these regulatory circuits. For example, previous studies have revealed that RctB can repress its own transcription [27], [28] as well as the transcription of rctA [28]. Transcription of rctA has been reported to inhibit the negative influence of rctA on RctB [8]. Additional regulatory processes also contribute to control of replication initiation. For example, ATP binding inhibits RctB activity by decreasing its ability to bind oriCII [7] and the methylation status of oriCII also influences RctB binding to oriCII [9]. Given the centrality of chromosome replication and segregation to the perpetuation of the species, the existence of multiple and perhaps redundant mechanisms to increase the robustness of the control of these processes is expected. Consistent with this idea, we only observed significant impairment of V. cholerae growth when RctB was over-expressed in an rctA mutant, a condition that likely allows considerable overinitiation of chrII. Overinitiation also leads to growth impairment and cell filamentation in E. coli and Caulobacter crescentus [29]–[31].
Although coordinated control of chromosome replication and segregation makes sense to ensure proper chromosome inheritance to daughter cells, little mechanistic information linking these essential processes is available. Recent work by Murray and colleagues revealed that in B. subtilis the ParA ortholog Soj can inhibit or stimulate chromosome replication initiation via interactions with the initiator protein DnaA, while the ParB ortholog Spo0J inhibits initiation of chromosome replication by blocking Soj dimerization [32], [33]. A similar regulatory scheme was recently described for V. cholerae chrI; Chattoraj and colleagues reported that ParA1 stimulates chrI replication and ParB1 inhibits ParA1 [34]. However, ParA2 appears to govern chrII replication initiation via a distinct mechanism that does not require it to interact with the replication initiator RctB. In contrast to findings for Soj and ParA1, which interact with DnaA, we did not detect interaction between ParA2 and RctB using a bacterial two hybrid system. Our findings, along with previous reports, suggest that further exploration of the roles of Par systems in control of chromosome replication in diverse bacteria is warranted. Since chromosomal par genes are found in ∼70% of bacterial genomes [20], Par proteins and parS sites may commonly exert control over chromosome replication. Finally, it will be interesting to explore whether mechanisms exist to link the replication and/or segregation of the two chromosomes in V. cholerae and other bacteria with multiple chromosomes.
Materials and Methods
Plasmids and strains
Most of the plasmids used in this study are listed in Table 2. The sites and mutations present in the rctA containing oriCII-based plasmids (discussed in Figure 1B[b]) are shown in Table S1. The plasmids used for the bacterial two hybrid analysis are shown in Table S2.

A two-step strategy for construction of oriCII-based plasmids was followed. First, different segments of DNA proximal to oriCII were amplified and cloned into pYB199, a derivative plasmid of pKD4 [35] which harbors the R6K origin and genes conferring resistance to ampicillin (bla) and kanamycin (aph). Second, the resulting plasmids were digested with XbaI and the fragment containing the oriCII region and aph was gel-purified, self-ligated and then electroporated into E. coli DH5α. Spontaneous suppressor mutants in the rctA containing oriCII-based plasmids (shown in Figure 2 and Table S1) were isolated by electroporating ∼100 ng of self-ligated DNA fragments into DH5α cells. Colonies that arose after an ∼48 hr incubation were re-streaked and then plasmid DNA was purified and sequenced. In addition, to confirm that mutation in the plasmid enabled establishment, each mutant plasmid was re-electroporated into DH5α. The mutations in parS2-B yielding parS2X (Figure 1A) were generated using the QuickChange XL Site Directed Mutagenesis Kit (Stratagene). To construct pYB141, pYB217, pYB447, and pYB448, 15-bp double stranded DNA fragments containing either parS2-A or parS2X were inserted into the EcoRI site of the vectors (pWKS30 and pXX705). Plasmids pYB193 and pYB216 were constructed in a similar fashion using the NheI and HindIII sites in the pBAD33 vector. To construct pYB451, the second copy of parS2-A was inserted at the AflII site of pYB447, which is ∼1.4 kb away from EcoRI site where the first copy of parS2-A was inserted. Plasmid pCB192-YY, a derivative of the transcriptional fusion vector pCB192 [36] in which the EcoRI site in the 3′ end of lacZ was removed by introduction of a silent mutation (GAATTC to GAATTT), was used to create transcriptional fusions to the parAB2 promoter. The parAB2 promoter region was amplified and cloned into the HindIII-EcoRI site of pCB192-YY. All the relevant DNA sequences of all plasmids used in this study were determined. The sequences of the oligonucleotides used in this study are listed in Dataset S1.
Mutations were introduced on to the V. cholerae chromosome (ΔrctA, and parS2-B::parS2X) via allele exchange using pCVD442-based plasmids as described [37]. V. cholerae strains used in this study are listed in Table 3.

Transformation efficiency experiments
DH5α cells harboring either pGZ119 (vector),or Isopropyl-β-D-1-thiogalactopyranoside (IPTG) inducible rctB or parB2, pYB285 or pYB273 respectively, were grown in LB broth containing 100 µM IPTG till mid-log phase to prepare electrocompetent cells. Similarly, DH5α cells harboring plasmid-borne parS2 sequences or control plasmids were grown until mid-log phase to prepare chemical competent cells. 40 ng of self-ligated DNA or 10 ng of pYB404 DNA were introduced into the competent cells. As a control, 10 ng of plasmid pYB190 was also introduced into competent cells in each experiment. The number of colonies obtained in the pYB190 transformations were used to normalize transformation efficiencies. Means and standard deviations were derived from 3–5 independent experiments for all plasmids tested.
Beta-galactosidase assays
Assays were performed in triplicate with log phase cultures as described previously [38]. Two tailed, two-sample equal t-tests were used to compare the results from 3 independent experiments (total 9 samples each) for the statistical analysis.
Electrophoretic mobility shift assay (EMSA)
Wild type RctB-His6, RctB[ΔC159]-His6, and ParB2-His6 proteins were purified as previously described [15]. Sequences used for oriCII, rctA, and rctA (parS2-B::parS2X) (see Figure 1A) EMSA probes were initially cloned into the pCR-Blunt II-TOPO vector (Invitrogen), yielding pYB405, pYB406, and pYB407, respectively. pYB190, a pCR-Blunt II TOPO derivative containing an irrelevant 10 bp, was used to construct the negative control probe. To prepare radio-labeled probes, appropriate DNA regions were amplified from the plasmids with universal M13 forward and reverse primers [22], end labeled with [γ-32P] ATP with polynucleotide kinase (New England Biolabs), purified from 6% DNA retardation gels (Invitrogen), and ethanol precipitated. In the binding reactions, 5,000 cpm of probe DNA containing different concentrations of RctB and/or ParB2 in a reaction buffer of 20 mM Tris-Cl (pH 7.5), 1 mM EDTA, 150 mM NaCl, 12.5 µg/mL poly (dI-dC), and 0.1 mg/mL BSA were incubated for 10 min at room temperature. The reactions were then electrophoresed in a 6% DNA retardation gel in 0.5× TAE and visualized by autoradiography.
DNase I foot print assay
DNase I footprint assays were performed as previously described with minor modifications [39]. The rctA probe was made by PCR using 5′-32P-radiolabeled rctA 5′-FP (CGTTTAAATAACCCACATATTCTTCGATAAGG) and rctA 3′-FP (ATACCTATTCGCTGGAGGAAAGATAGG) primers on a plasmid encoding parAB2-rctA-oriCII-rctB (pYB403). The probe was purified from 6% DNA retardation gels, eluted, and ethanol precipitated. 1,200,000 cpm of probe was incubated with different amounts of RctB without and with 100 ng of ParB2 in 20 µL of 20 mM Tris-Cl pH 8.0, 125 mM NaCl, 1 mM DTT for 10 min at room temperature. 0.1 U of DNase I (Applied Biosystems) was added to each reaction and incubated at room temperature for 30 sec. The digestions were quenched by the addition of 6 µL of 660 mM Tris-Cl pH 9.5, 66 mM EDTA, 3.3% SDS and placed on ice. Samples were ethanol precipitated, resuspended in recrystalized formamide, and 20,000 cpm of each was run on an 8% polyacrylamide gel with 8 M urea (National Diagnostics) in 1× TBE. The gels were then dried and visualized by autoradiography.
Quantitative PCR assay
Genomic DNA was prepared from each strain by phenol-chloroform extraction followed by ethanol precipitation. The genomic DNA was then digested with PstI and 10 pg was used for each quantitative PCR (qPCR) reaction. Genomic DNA from an N16961 stationary culture was used to generate the standard curve. qPCR was performed with the StepOnePlus Real-Time PCR system (Applied Biosciences) using SYBR Green Master mix (Applied Biosciences) according to the manufacturer's protocol. The primer pairs used for oriCI and oriCII were described previously [4]. Each qPCR run was done in triplicate and the ratio was calculated from three independent experiments.
Growth curves and microscopy
V. cholerae cells harboring a plasmid borne copy of an IPTG-inducible rctB or a control vector were grown in either LB or M9-Glucose media containing 100 µM IPTG at starting OD600 of 0.003. Subsequently, OD600 and CFU were monitored hourly. Growth curves shown in Figure S2 are representative of at least three independent experiments. A small aliquot of cells was removed at 4 hr, fixed with 3% paraformaldehyde, and then examined with 100× alpha-plan lens on a Zeiss Axioplan 2 microscope.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. OkadaKIidaTKita-TsukamotoKHondaT 2005 Vibrios commonly possess two chromosomes. J Bacteriol 187 752 757
2. HeidelbergJFEisenJANelsonWCClaytonRAGwinnML 2000 DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406 477 483
3. EganESWaldorMK 2003 Distinct replication requirements for the two Vibrio cholerae chromosomes. Cell 114 521 530
4. DuigouSKnudsenKGSkovgaardOEganESLøbner-OlesenA 2006 Independent control of replication initiation of the two Vibrio cholerae chromosomes by DnaA and RctB. J Bacteriol 188 6419 6424
5. MottMLBergerJM 2007 DNA replication initiation: mechanisms and regulation in bacteria. Nat Rev Microbiol 5 343 354
6. Zakrzewska-CzerwińskaJJakimowiczDZawilak-PawlikAMesserW 2007 Regulation of the initiation of chromosomal replication in bacteria. FEMS Microbiol Rev 31 378 387
7. DuigouSYamaichiYWaldorMK 2008 ATP negatively regulates the initiator protein of Vibrio cholerae chromosome II replication. Proc Natl Acad Sci U S A 105 10577 10582
8. Venkova-CanovaTSrivastavaPChattorajDK 2006 Transcriptional inactivation of a regulatory site for replication of Vibrio cholerae chromosome II. Proc Natl Acad Sci U S A 103 12051 12056
9. DemarreGChattorajDK 2010 DNA adenine methylation is required to replicate both Vibrio cholerae chromosomes once per cell cycle. PLoS Genet 6 e1000939 doi:10.1371/journal.pgen.1000939
10. EganESLøbner-OlesenAWaldorMK 2004 Synchronous replication initiation of the two Vibrio cholerae chromosomes. Curr Biol 14 R501 R502
11. RasmussenTJensenRBSkovgaardO 2007 The two chromosomes of Vibrio cholerae are initiated at different time points in the cell cycle. EMBO J 26 3124 3131
12. StokkeCWaldminghausTSkarstadK 2011 Replication patterns and organization of replication forks in Vibrio cholerae. Microbiology 157 695 708
13. FogelMAWaldorMK 2006 A dynamic, mitotic-like mechanism for bacterial chromosome segregation. Genes Dev 20 3269 3282
14. Saint-DicDFrushourBPKehrlJHKahngLS 2006 A parA homolog selectively influences positioning of the large chromosome origin in Vibrio cholerae. J Bacteriol 188 5626 5631
15. YamaichiYFogelMAMcLeodSMHuiMPWaldorMK 2007 Distinct centromere-like parS sites on the two chromosomes of Vibrio spp. J Bacteriol 189 5314 5324
16. YamaichiYFogelMAWaldorMK 2007 par genes and the pathology of chromosome loss in Vibrio cholerae. Proc Natl Acad Sci U S A 104 630 635
17. GerdesKHowardMSzardeningsF 2010 Pushing and pulling in prokaryotic DNA segregation. Cell 141 927 942
18. SaljeJ 2010 Plasmid segregation: how to survive as an extra piece of DNA. Crit Rev Biochem Mol Biol 45 296 317
19. LinDCGrossmanAD 1998 Identification and characterization of a bacterial chromosome partitioning site. Cell 92 675 685
20. LivnyJYamaichiYWaldorMK 2007 Distribution of centromere-like parS sites in bacteria: insights from comparative genomics. J Bacteriol 189 8693 8703
21. YamaichiYDuigouSShakhnovichEAWaldorMK 2009 Targeting the Replication Initiator of the Second Vibrio Chromosome: Towards Generation of Vibrionaceae-Specific Antimicrobial Agents. PLoS Pathog 5 e1000663 doi:10.1371/journal.ppat.1000663
22. SambrookJRussellD 2001 Molecular Cloning. Cold Spring Habor Laboratory Press
23. SrivastavaPChattorajDK 2007 Selective chromosome amplification in Vibrio cholerae. Mol Microbiol 66 1016 1028
24. de la HozABAyoraSSitkiewiczIFernándezSPankiewiczR 2000 Plasmid copy-number control and better-than-random segregation genes of pSM19035 share a common regulator. Proc Natl Acad Sci U S A 97 728 733
25. CarmeloEBarillàDGolovanovAPLianLYDeromeA 2005 The unstructured N-terminal tail of ParG modulates assembly of a quaternary nucleoprotein complex in transcription repression. J Biol Chem 280 28683 28691
26. RinggaardSEbersbachGBorchJGerdesK 2007 Regulatory cross-talk in the double par locus of plasmid pB171. J Biol Chem 282 3134 3145
27. PalDVenkova-CanovaTSrivastavaPChattorajDK 2005 Multipartite regulation of rctB, the replication initiator gene of Vibrio cholerae chromosome II. J Bacteriol 187 7167 7175
28. EganESDuigouSWaldorMK 2006 Autorepression of RctB, an initiator of Vibrio cholerae chromosome II replication. J Bacteriol 188 789 793
29. KatayamaTTakataMSekimizuK 1997 CedA is a novel Escherichia coli protein that activates the cell division inhibited by chromosomal DNA over-replication. Mol Microbiol 26 687 697
30. FujimitsuKSu'etsuguMYamaguchiYMazdaKFuN 2008 Modes of overinitiation, dnaA gene expression, and inhibition of cell division in a novel cold-sensitive hda mutant of Escherichia coli. J Bacteriol 190 5368 5381
31. CollierJShapiroL 2009 Feedback control of DnaA-mediated replication initiation by replisome-associated HdaA protein in Caulobacter. J Bacteriol 191 5706 5716
32. MurrayHErringtonJ 2008 Dynamic control of the DNA replication initiation protein DnaA by Soj/ParA. Cell 135 74 84
33. ScholefieldGWhitingRErringtonJMurrayH 2011 Spo0J regulates the oligomeric state of Soj to trigger its switch from an activator to an inhibitor of DNA replication initiation. Mol Microbiol 79 1089 1100
34. KadoyaRBaekJHSarkerAChattorajDK 2011 Participation of a Chromosome Segregation Protein ParAI of Vibrio cholerae in Chromosome Replication. J Bacteriol 193 1504 1514
35. DatsenkoKAWannerBL 2000 One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97 6640 6645
36. SchneiderKBeckCF 1986 Promoter-probe vectors for the analysis of divergently arranged promoters. Gene 42 37 48
37. DonnenbergMSKaperJB 1991 Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect Immun 59 4310 4317
38. MillerJH 1992 A short course in bacterial genetics Cold Spring Habor Laboratory Press
39. BruistMFGlasgowACJohnsonRCSimonMI 1987 Fis binding to the recombinational enhancer of the Hin DNA inversion system. Genes Dev 1 762 772
40. GuzmanLMBelinDCarsonMJBeckwithJ 1995 Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177 4121 4130
41. LesslMBalzerDLurzRWatersVLGuineyDG 1992 Dissection of IncP conjugative plasmid transfer: definition of the transfer region Tra2 by mobilization of the Tra1 region in trans. J Bacteriol 174 2493 2500
42. WangRFKushnerSR 1991 Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100 195 199
43. NikiHHiragaS 1997 Subcellular distribution of actively partitioning F plasmid during the cell division cycle in E. coli. Cell 90 951 957
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 7
Nejčtenější v tomto čísle
- A Rice Plastidial Nucleotide Sugar Epimerase Is Involved in Galactolipid Biosynthesis and Improves Photosynthetic Efficiency
- Genome-Wide Association Study Identifies Novel Restless Legs Syndrome Susceptibility Loci on 2p14 and 16q12.1
- Loss of the BMP Antagonist, SMOC-1, Causes Ophthalmo-Acromelic (Waardenburg Anophthalmia) Syndrome in Humans and Mice
- Gene-Based Tests of Association