
Germline Variation Controls the Architecture of Somatic Alterations in Tumors
Studies have suggested that somatic events in tumors can depend on an individual's constitutional genotype. We used squamous cell carcinomas (SCC) of the skin, which arise in high multiplicity in organ transplant recipients, as a model to compare the pattern of somatic alterations within and across individuals. Specifically, we performed array comparative genomic hybridization on 104 tumors from 25 unrelated individuals who each had three or more independently arisen SCCs and compared the profiles occurring within patients to profiles of tumors across a larger set of 135 patients. In general, chromosomal aberrations in SCCs were more similar within than across individuals (two-sided exact-test p-value ), consistent with the notion that the genetic background was affecting the pattern of somatic changes. To further test this possibility, we performed allele-specific imbalance studies using microsatellite markers mapping to 14 frequently aberrant regions of multiple independent tumors from 65 patients. We identified nine loci which show evidence of preferential allelic imbalance. One of these loci, 8q24, corresponded to a region in which multiple single nucleotide polymorphisms have been associated with increased cancer risk in genome-wide association studies (GWAS). We tested three implicated variants and identified one, rs13281615, with evidence of allele-specific imbalance (p-value = 0.012). The finding of an independently identified cancer susceptibility allele with allele-specific imbalance in a genomic region affected by recurrent DNA copy number changes suggest that it may also harbor risk alleles for SCC. Together these data provide strong evidence that the genetic background is a key driver of somatic events in cancer, opening an opportunity to expand this approach to identify cancer risk alleles.
Published in the journal:
. PLoS Genet 6(9): e32767. doi:10.1371/journal.pgen.1001136
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001136
Summary
Studies have suggested that somatic events in tumors can depend on an individual's constitutional genotype. We used squamous cell carcinomas (SCC) of the skin, which arise in high multiplicity in organ transplant recipients, as a model to compare the pattern of somatic alterations within and across individuals. Specifically, we performed array comparative genomic hybridization on 104 tumors from 25 unrelated individuals who each had three or more independently arisen SCCs and compared the profiles occurring within patients to profiles of tumors across a larger set of 135 patients. In general, chromosomal aberrations in SCCs were more similar within than across individuals (two-sided exact-test p-value ), consistent with the notion that the genetic background was affecting the pattern of somatic changes. To further test this possibility, we performed allele-specific imbalance studies using microsatellite markers mapping to 14 frequently aberrant regions of multiple independent tumors from 65 patients. We identified nine loci which show evidence of preferential allelic imbalance. One of these loci, 8q24, corresponded to a region in which multiple single nucleotide polymorphisms have been associated with increased cancer risk in genome-wide association studies (GWAS). We tested three implicated variants and identified one, rs13281615, with evidence of allele-specific imbalance (p-value = 0.012). The finding of an independently identified cancer susceptibility allele with allele-specific imbalance in a genomic region affected by recurrent DNA copy number changes suggest that it may also harbor risk alleles for SCC. Together these data provide strong evidence that the genetic background is a key driver of somatic events in cancer, opening an opportunity to expand this approach to identify cancer risk alleles.
Introduction
Human solid cancers are characterized by the presence of numerous genetic alterations that accumulate during the evolution of the disease. While the mutation spectrum within biologically related cancer subtypes often shows similarities with regards to the patterns of genetic alteration, each individual cancer has a unique combination of alterations. The forces that shape the genomic landscape of individual cancers are in part determined by the nature of the initiating oncogenic alterations and the sequence in which they occur. However, the constitutional genotype of the cell acquiring the first pathogenetically relevant mutation is likely to play a role in influencing which somatic alterations will undergo positive or negative selection. The influence of inherited alterations on the pattern of somatic mutations found in evolved cancers has been demonstrated in several cancer types. In breast cancers from individuals with inherited BRCA1 mutations one finds more frequent losses on 4p, 4q, 5q, Xp and Xq and gains of 10p and 16q compared to breast tumors from individuals without BRCA1 mutations [1], [2]. In melanoma, patients with germline variations in MC1R have a higher frequency of somatic BRAF mutations in their melanomas than patients without MC1R variants [3], [4]. These examples of interactions between predisposing germline alterations and acquired mutations in the tumor occur between different genes (trans-effects). Several studies have also identified cis-effects, in which somatic alterations affect specific inherited variants. Examples include two genes identified through mouse mapping studies: AURKA, which shows allele-specific gains of the T91A allele in human colon tumors [5], [6] and PTPRJ, which shows allele-specific losses of the A1176C allele in human colon tumors [7]. In addition, rs6983267, a SNP on 8q24 found through several genome-wide association studies to be associated with susceptibility to colorectal cancer, shows allele-specific imbalance [8].
Together these data suggest that inherited variation as well as somatic mutations arising early in progression help shape the pattern of somatic changes that occur subsequently during tumor evolution. One way to more systematically assess the effect of the constitutional genotype on the pattern of somatic alterations is to compare cancers of the same type that arose independently on the common genetic background of a single individual. Only a few cancer types arise frequently enough to render such analysis practical. Basal cell carcinoma and squamous cell carcinoma (SCC) of the skin often develop in multiplicity. Furthermore, the incidence of SCC in particular is dramatically increased in immunosuppressed patients. Specifically, in organ transplant recipients (OTRs) the risk of SCC is 65 to 250 fold increased compared to the general population [9]. As a consequence, some patients develop dozens of separate primary carcinomas. In this study, we exploited the unique property of the OTR population to test the hypothesis that tumors arising on a common genetic background will have somatic alterations that are more similar to each other than to those found in similar tumors that developed in different individuals and whether this scenario can be exploited to discover predisposing genetic factors.
Results
We obtained copy number profiles as measured by array comparative genomic hybridization (aCGH) from tumors arising in individuals with multiple independent cutaneous squamous cell carcinomas (SCCs) or keratocanthomas (KAs) (intra-group) and copy number profiles of SCCs and KAs of separate individuals (inter-group). 305 independent tumor samples from 181 patients were included in this initial study (Figure 1). As previously reported [10], [11], focal genomic aberrations were rare in these tumors and DNA copy number aberrations consisted mostly of the loss or gain of whole chromosome arms. As the resolution of copy number changes using aCGH is around 1Mb, it is possible that we missed focal amplifications or deletions in this study.
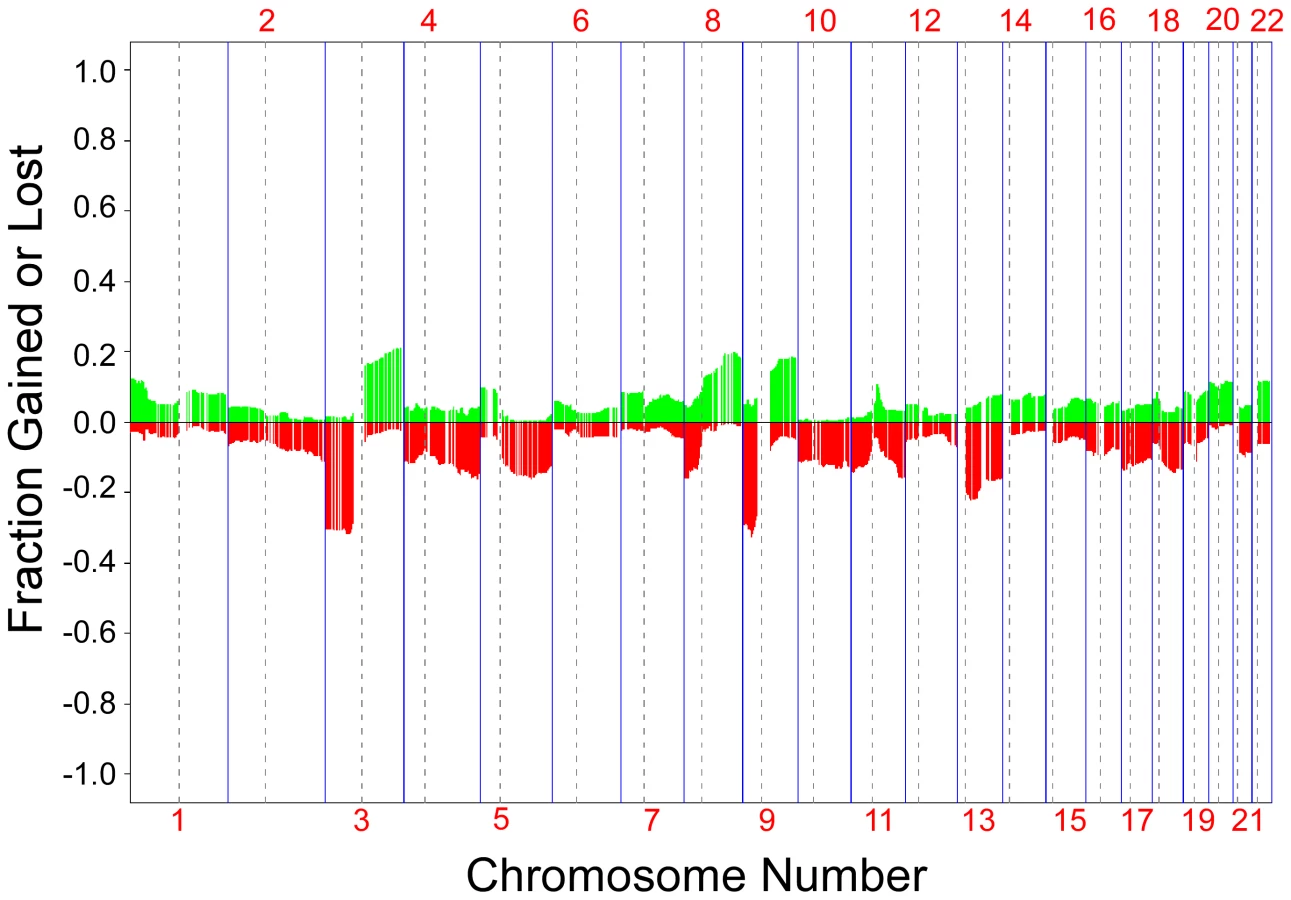
We compared aCGH profiles between the three types of skin tumors in our study, SCC, SCC in situ (Bowen's Disease) and keratocanthoma. There were no statistically significant differences in frequency of clone loss or gain between the SCC and keratocanthoma profiles; however there were several loci which showed differences between the SCC in situ profiles and profiles from the other two tumor types (data not shown). Because of this, we focused our comparative analysis on SCCs only. Our data set included 222 SCCs from 135 individuals. From 25 of those individuals, three or more SCCs (median = 4.2; range 3–6) were analyzed to compare the intra-group and inter-group similarities of DNA copy number changes.
We found a significantly higher concordance of chromosomal aberrations in SCCs within than between patients [two-sided T-test p-values: 6.97×10−8](Figure 2). Interestingly, certain chromosomal regions (4q, 11q, and 17q) were preferentially affected by this concordance (individual arm p-values<0.05; Table 1). The intra-group correlation coefficients (ICC) for the array elements of these regions were compared but did not allow narrowing the genomic region to specific loci within these regions. This is not unexpected, considering that most of the tumors showed copy number changes affecting large genomic regions, such as entire chromosomal arms or chromosomes.
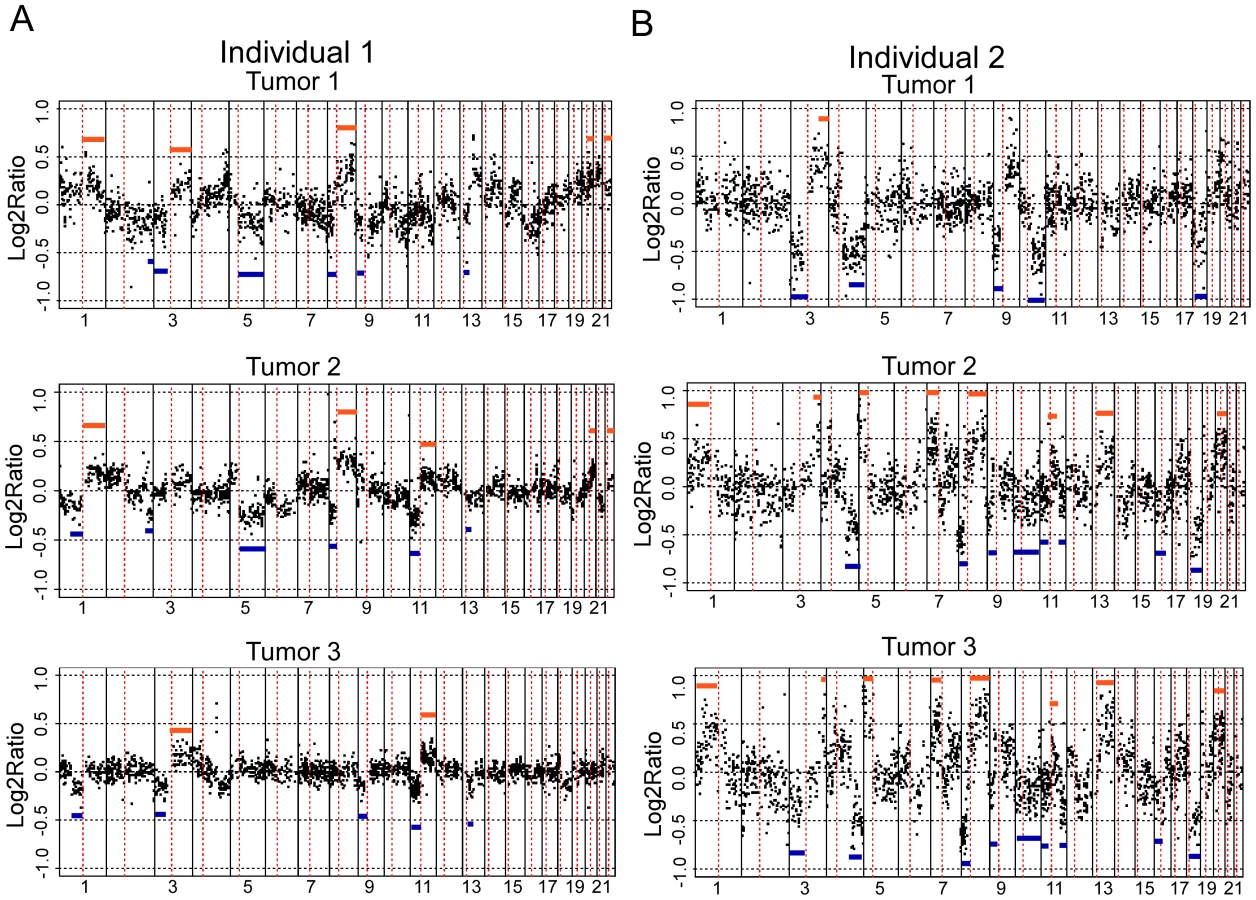
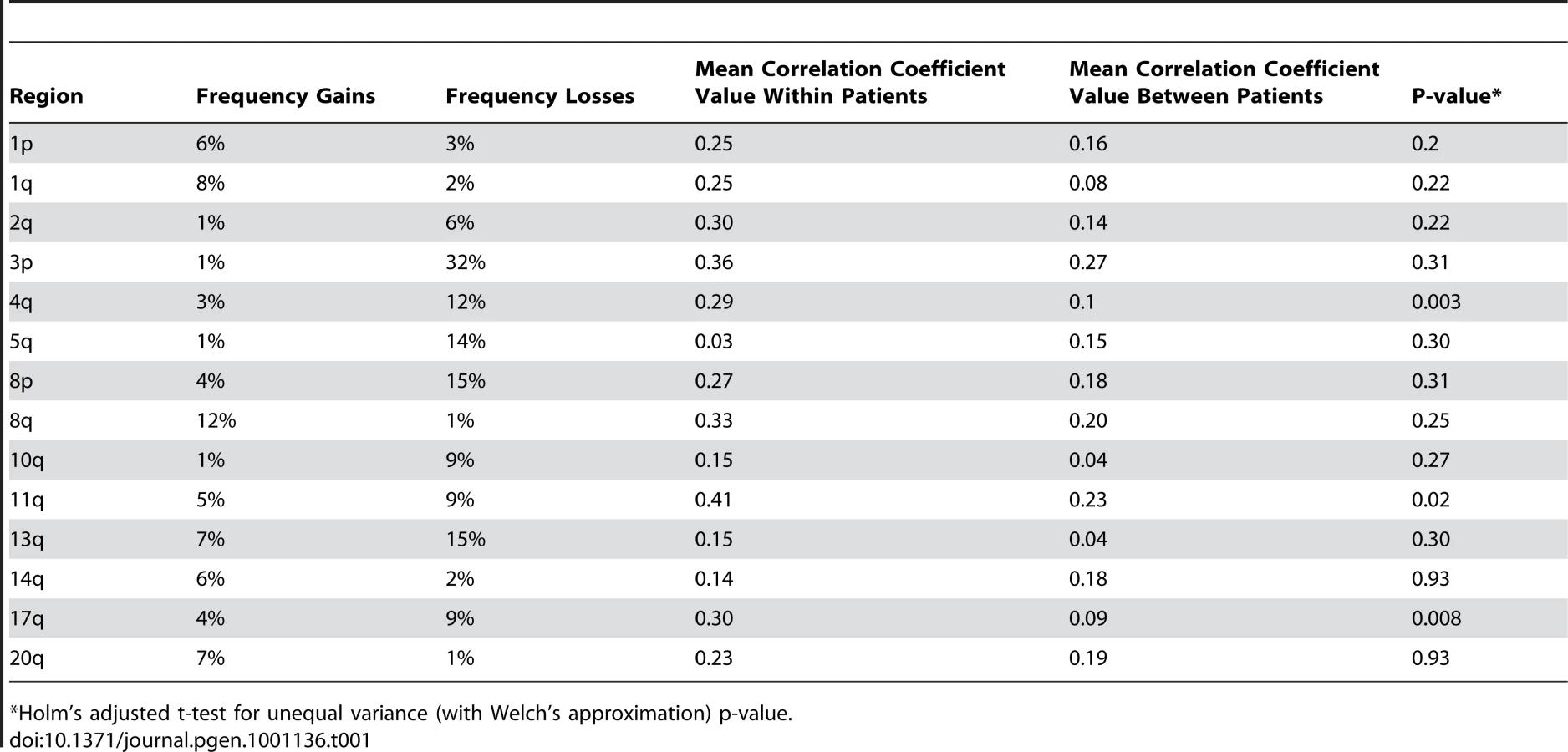
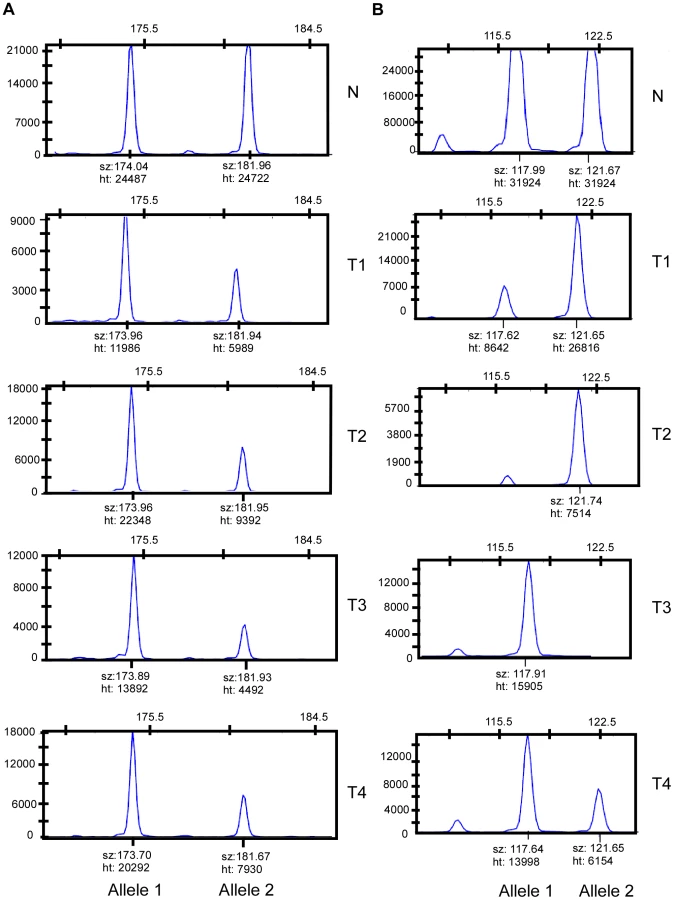
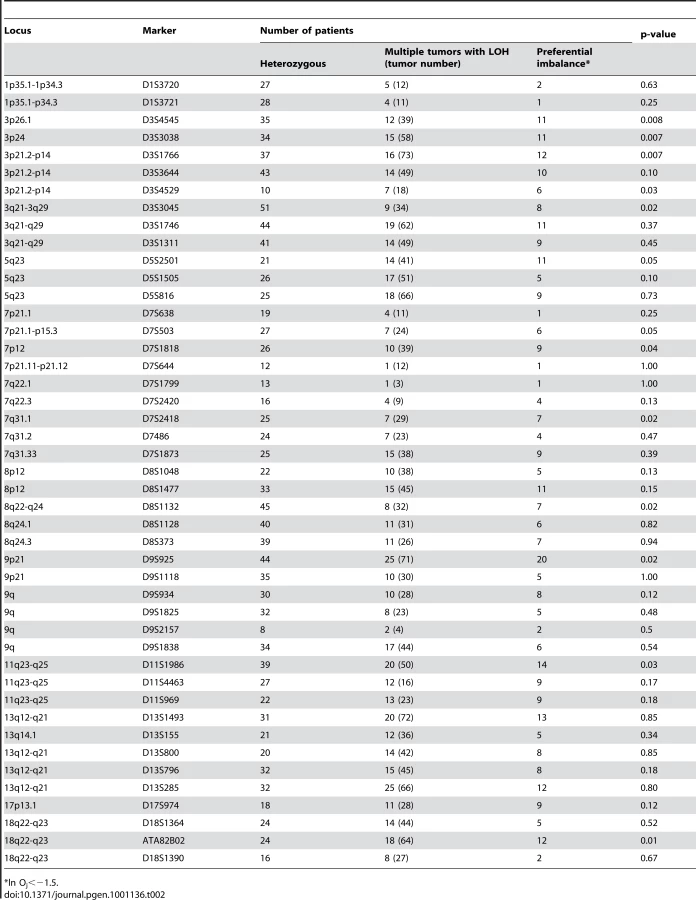
We rationalized that any inherited variants that promote cancer in an allele-specific manner would result in allele-specific DNA copy number changes reflected by preferential loss or gain of one specific chromosome in the tumors of an individual patient. By contrast, dosage events affecting genes that promote cancer in allele-independent manner, e.g. loss of CDKN2A or gain of MYC, were expected to display random somatic alterations of either allele [12]. To determine the presence of allele-specific changes occurring within tumors of individual patients, we performed loss of heterozygosity analyses of 45 microsatellite markers covering 14 chromosomal regions that were chosen based on the frequency of aberration as measured by aCGH and without prior knowledge of regions showing more similarity within versus across patients. 270 tumors from 65 individuals with a minimum of three independent tumors were included in this analysis. The constitutional genotype was determined from DNA extracted from blood leukocytes of each patient. Allelic imbalance was defined as a tumor to normal DNA allelic ratio of greater than 1.5 or less than 0.67. Statistical analyses for preferential imbalance were conducted for individuals who were heterozygous for a given marker and had two or more tumors showing imbalance; two examples are illustrated in Figure 3. Thirteen markers representing eight different genomic regions showed significant skewing towards one allele as determined by a Bayesian/frequentist approach (Text S1). Markers demonstrating significant preferential allelic imbalance mapped to chromosomal locations 3p24, 3q21-26, 5q23, 7p12-21, 7q31, 8q24, 9p21, 11q24, and 18q22 (Table 2, Figure 3). These data indicate that the increased similarity of copy number changes within individuals is at least in part due to inherited variation within the same region as the copy number change.
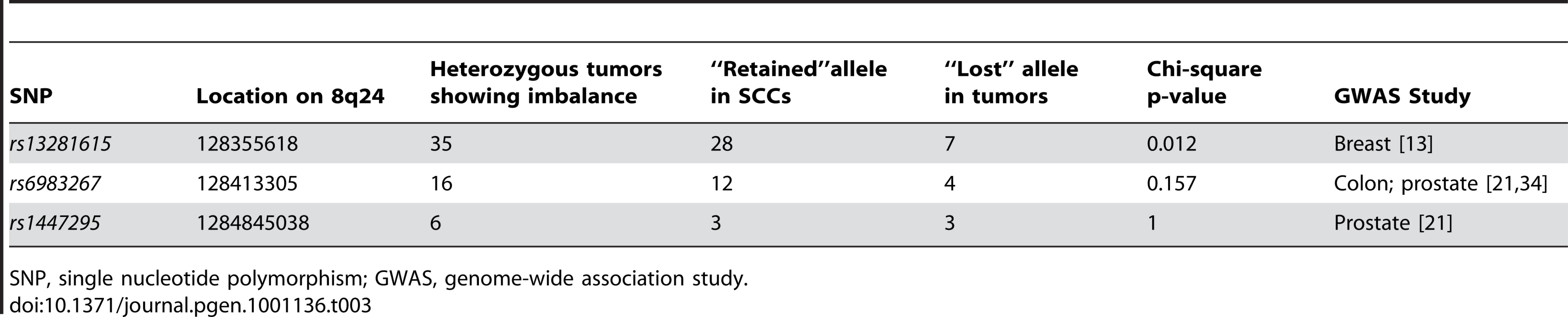
The next question we addressed was whether variations in any known tumor susceptibility genes were driving allele-specific imbalance at the loci identified through our studies. Several genome-wide association studies (GWAS) have been performed for multiple cancers including breast, prostate, colon, and melanoma [13]–[15]. Variants at 8q24 identified via GWAS have been associated for cancer risk for multiple cancer types [8], [13], [16]–[21]. To determine if any of these were candidates for the observed allele-specific imbalances at 8q24, we tested three variants, rs13281615, rs1447295, and rs6983267, for allele-specific imbalances in matched normal and tumor DNAs from individuals with SCC. Of these, only rs13281615 showed statistically significant evidence of allelic skewing (Table 3). Of 35 heterozygous tumors showing imbalance for rs13281615, 28 of them showed an imbalance in favor of the A allele while only 7 showed an imbalance favoring the G allele (p-value 0.012). A second SNP, rs6983267, showed a similar trend that did not reach statistical significance (p-value 0.157). These data raise the possibility that rs13291615 may be a candidate susceptibility allele for SCC. Our results suggest that the use of preferential allelic imbalance may be an efficient approach to map susceptibility variants in specific clinical settings.
Discussion
In summary, our finding of an increased concordance of DNA copy number changes together with the presence of allelic-specific imbalance within separate cancers of individuals strongly suggests that the somatic changes occurring in tumors are in part affected by underlying characteristics of the individual host. An in depth comparison of allele-specific genomic changes occurring in multiple tumors of individual patients may offer a unique route to uncover cancer susceptibility loci.
The allele-specific LOH data from both microsatellite analysis and from SNP analysis indicate that the increased similarity of copy number changes within individuals is at least in part due to inherited variation within the same region as the copy number change. By contrast, not all loci that were frequently affected by concordant aberrations within individuals showed evidence of preferential allelic imbalance. This could be due to trans-effects between inherited variants elsewhere in the genome and a cancer gene in the region affected by the copy number alteration. For example, 13q12-q21, containing the Rb tumor suppressor gene, showed frequent loss in SCCs and a high intra-group concordance but did not show evidence of preferential imbalance.
There are some alternative explanations for the greater similarity of changes in tumors within versus between individuals. In our study we defined tumors as being independent based on arising in different anatomical sites. This should reduce the probability that tumors are related via a shared clonal origin. It is unlikely, but not impossible that tumors arising on different sites might have a common precursor which would explain the results of this study. It also remains possible that other pathogenetic factors such as ultraviolet light exposure or the presence of human papilloma virus may also influence the similarity of somatic alterations of tumors arising within an individual that do not show allele-specific imbalance. Finally, different immunosuppressive drugs may result in specific mutations occurring in tumors which might manifest as similar copy number patterns in tumors from within an individual. Another explanation of our results is that environmental exposures may result in differential selection between alleles which could result in allele-specific imbalances. Despite these possibilities, our study strongly supports the notion that the constitutional genotype of an individual exerts a strong influence on the somatic alterations that arise in cancer. Genetic analyses of cancer that arise at high multiplicity may offer a novel route to the discovery of cancer susceptibility genes.
Methods
Human samples
All study participants signed informed consent and the study was approved by University of California San Francisco (UCSF) and Ohio State University (OSU) Institutional Review Boards. Participants were eligible if they had available SCC and normal tissue available for study. To reduce the possibility that tumors from the same individual might be related clonally, we chose tumors from different anatomical locations when they were excised on the same day. Tumors excised on different dates also needed to be excised from different anatomical locations. Re-excisions were not included in the study. Tumor DNA was microdissected from formalin-fixed paraffin embedded tissue sections containing at least 70% tumor cells and the concentration was measured using TaqMan analysis [22]. Blood DNA was used as a source of normal reference DNA for loss of heterozygosity analyses.
aCGH
We obtained aCGH profiles from a total of 305 tumors from 181 patients and these consisted of one actinic keratosis, 37 Bowen's disease, 45 KAs and 222 SCCs. We focused our subsequent analyses on 222 SCCs from 135 individuals. The cohort included 25 patients who had 3 or more independent tumors that were examined by aCGH (number of tumors, n = 104). Tumor genomic DNA (1000ng) and reference DNA (600ng) (Promega) was labeled with Cy3 and Cy5, respectively using random primers essentially as previously described [23], [24]. The labeled tumor and reference DNA was pooled and applied to Hum3.2 BAC arrays for 48 hours. The arrays contained 2464 BAC clones with an average resolution of 100 Mb. Analysis of the arrays was carried out using R/Bioconductor software [25], [26]. Prior to analysis the data was normalized with respect to GC content and geometrical position on the arrays [27]. Regions of equal copy number were defined by segmenting the data using circular binary segmentation (CBS) [28]. The scaled median absolute deviation (MAD) of the difference between the observed and segmented values was used to estimate the sample-specific experimental variation; samples with a MAD of greater than 0.2 (n = 18) were considered unsuitable for inclusion in the study. The gain and loss status for each probe was defined using the merged level procedure [29].
Statistical analyses
For each autosomal arm, correlation coefficients based on log2 ratio values were computed for each pair of samples for those patients who had at least three independent SCC samples. Only those sample pairs were considered where at least one of the samples had 20% of clones with absolute value greater than 2 times sample MAD and another 20% below 2 times MAD. This ensured that the correlation was not driven by a flat sample profile. Only those arms were considered where there were at least 40 clones with non-missing values and at least 20 sample pairs in each of intra and inter groups; arms excluded from analyses include 2p, 3q, 4p, 5p, 6p, 6q, 7p, 7q, 9p, 9q, 10p, 10q, 11p, 12p, 12q, 15q, 16p, 16q, 17p, 18p, 18q, 19p, 19q, 20p, 21q and 22q. Two-sided, two sample t-tests were performed comparing the intra and inter patient groups for each of those arms where there was no significant difference in group variances. Brown-Forsythe version of the Levene-type test [30] was used to test for unequal group variances. The t-test p-values were then adjusted for multiple testing by Holm's method. Since a number of arms had unequal group variances, t-tests with Welch's approximation were also performed on each arm. Theoretical p-values were then adjusted by Holm's method. Genome-wide p-value was similarly computed by considering clones from all autosomes. There was no difference in group variances when considering whole genome. The correlation coefficients when considering individual arms and also whole genome had near normal distributions.
We identified clones having high within vs. between patient effects by estimating intraclass correlation coefficient (ICC) which captured the within-patient similarity. A random effects model Yij = μ + αi + εij, where the response variable is the CBS value with original log2ratio if a clone is an outlier in that segment, j and i represent the tumor and patient respectively, μ is an unobserved overall mean, αi is an unobserved random effect shared by all tumors in patient i, and εij is an unobserved noise term, was fit for each clone and ICC calculated as σα2/(σα2+σε2) where σα2 and σε2 are the variances of αi and εij respectively.
Microsatellite LOH analyses
Matched normal and tumor DNAs from 65 individuals were genotyped for 45 microsatellite markers mapping to regions of common chromosomal loss or gain. A total of 270 skin tumors were studied for microsatellite LOH analyses. For allelotyping, we chose microsatellite markers with a high degree of heterozygosity that can be readily quantified. To allow efficient amplification from fixed tissue we selected microsatellite markers with PCR product sizes less than 200 bp [12]. Fluorescently labeled, multiplexed PCR products were analyzed on an ABI 377 DNA sequencer using GeneMapper v3.7 (Applied Biosystems) in the OSU Comprehensive Cancer Center Nucleic Acids Shared Resource. An allelic imbalance ratio (R) in each tumor sample for each marker was calculated using a standard protocol: R = (TA/TB)/(NA/NB), where TA is a peak height from tumor DNA of the larger sized allele, TB is the peak height area from tumor DNA of the smaller sized allele, NA is the peak height area from normal DNA of the larger allele, and NB is the peak height area from normal DNA of the smaller allele. As described by others, when R was greater than 1.5 or less than 0.66, the sample was considered to have allelic imbalance [31], [32]. When R was between 1.25 and 0.85 the sample was considered to have no imbalance. Other values for R were treated as uncertain.
Preferential imbalance analyses of microsatellite genotypes
Allelic imbalance data using microsatellite markers cannot simply be compiled across individuals, due to the heterozygosity of allele sizes across individuals each individual is likely to have a different combination of genotypes. We used a Bayesian/frequentist approach which we developed specifically for these data to determine if any given marker showed preferential allelic imbalance within and across patients (Text S1). In brief, we evaluated patient-specific odds of preferential imbalance as an indicator of randomness in a given individual using a Bayesian method. We then combined these odds into a sample to assess the evidence in favor of preferential imbalance in the general population using a frequentist method. A Wilcoxon rank sum test was then performed and all loci that rejected the null hypothesis at a 5% level of significance were deemed to show preferential imbalance.
Allele-specific imbalance studies
We conducted quantitative genotyping of matched normal and SCC tumor DNA pairs using Sequenom MassARRAY Iplex gold genotyping technology. It is highly quantitative and is extremely sensitive for detection of allelic gains or losses in tumors and has been used for allelic imbalance studies [33]. All genotypes of poorer quality (aggressive calls) and those for whom a water sample had a strong call were eliminated from further analysis. Genotypes were also discarded from analysis if one of the two paired normal/tumor DNAs did not work resulting in genotypes included in analysis from 299 SCCs from 130 individuals for rs13281615, 110 SCCs for 70 individuals for rs1447295, and 175 SCCs from 84 individuals for rs6983267. Genotypes and peak area data for each allele were analyzed to identify regions of genomic imbalance between each matched normal and tumor DNA. An allelic imbalance ratio (R) to measure imbalance in each tumor sample for each SNP was calculated as described for the microsatellite LOH studies. Duplicate SNPs were included for quality control and two control samples and two no template controls were used. Chi-squared analyses were used to determine significance of observed allelic imbalances compared to expected 50∶50 imbalances indicative of random allelic imbalance.
Supporting Information
Zdroje
1. JonssonG
NaylorTL
Vallon-ChristerssonJ
StaafJ
HuangJ
2005 Distinct genomic profiles in hereditary breast tumors identified by array-based comparative genomic hybridization. Cancer Res 65 7612 7621
2. StefanssonOA
JonassonJG
JohannssonOT
OlafsdottirK
SteinarsdottirM
2009 Genomic profiling of breast tumours in relation to BRCA abnormalities and phenotypes. Breast Cancer Res 11 R47
3. LandiMT
BauerJ
PfeifferRM
ElderDE
HulleyB
2006 MC1R germline variants confer risk for BRAF-mutant melanoma. Science 313 521 522
4. MaldonadoJL
FridlyandJ
PatelH
JainAN
BusamK
2003 Determinants of BRAF mutations in primary melanomas. J Natl Cancer Inst 95 1878 1890
5. Ewart-TolandA
BriassouliP
de KoningJP
MaoJH
YuanJ
2003 Identification of Stk6/STK15 as a candidate low-penetrance tumor-susceptibility gene in mouse and human. Nat Genet 34 403 412
6. HienonenT
SalovaaraR
MecklinJP
JarvinenH
KarhuA
2006 Preferential amplification of AURKA 91A (Ile31) in familial colorectal cancers. Int J Cancer 118 505 508
7. RuivenkampCA
van WezelT
ZanonC
StassenAP
VlcekC
2002 Ptprj is a candidate for the mouse colon-cancer susceptibility locus Scc1 and is frequently deleted in human cancers. Nat Genet 31 295 300
8. TuupanenS
NiittymakiI
NousiainenK
VanharantaS
MecklinJP
2008 Allelic imbalance at rs6983267 suggests selection of the risk allele in somatic colorectal tumor evolution. Cancer Res 68 14 17
9. DiGiovannaJJ
1998 Posttransplantation skin cancer: scope of the problem, management, and role for systemic retinoid chemoprevention. Transplant Proc 30 2771 2775; discussion 2776–2778
10. AshtonKJ
WeinsteinSR
MaguireDJ
GriffithsLR
2003 Chromosomal aberrations in squamous cell carcinoma and solar keratoses revealed by comparative genomic hybridization. Arch Dermatol 139 876 882
11. PoppS
WalteringS
Holtgreve-GrezH
JauchA
ProbyC
2000 Genetic characterization of a human skin carcinoma progression model: from primary tumor to metastasis. J Invest Dermatol 115 1095 1103
12. MaoX
HamoudiRA
TalbotIC
BaudisM
2006 Allele-specific loss of heterozygosity in multiple colorectal adenomas: toward an integrated molecular cytogenetic map II. Cancer Genet Cytogenet 167 1 14
13. EastonDF
PooleyKA
DunningAM
PharoahPD
ThompsonD
2007 Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 447 1087 1093
14. FalchiM
BatailleV
HaywardNK
DuffyDL
BishopJA
2009 Genome-wide association study identifies variants at 9p21 and 22q13 associated with development of cutaneous nevi. Nat Genet 41 915 919
15. TenesaA
FarringtonSM
PrendergastJG
PorteousME
WalkerM
2008 Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat Genet 40 631 637
16. ChengI
PlummerSJ
JorgensonE
LiuX
RybickiBA
2008 8q24 and prostate cancer: association with advanced disease and meta-analysis. Eur J Hum Genet 16 496 505
17. FletcherO
JohnsonN
GibsonL
CouplandB
FraserA
2008 Association of genetic variants at 8q24 with breast cancer risk. Cancer Epidemiol Biomarkers Prev 17 702 705
18. Garcia-ClosasM
HallP
NevanlinnaH
PooleyK
MorrisonJ
2008 Heterogeneity of breast cancer associations with five susceptibility loci by clinical and pathological characteristics. PLoS Genet 4 e1000054
19. LiL
PlummerSJ
ThompsonCL
MerkulovaA
AchesonLS
2008 A common 8q24 variant and the risk of colon cancer: a population-based case-control study. Cancer Epidemiol Biomarkers Prev 17 339 342
20. WokolorczykD
GliniewiczB
SikorskiA
ZlowockaE
MasojcB
2008 A range of cancers is associated with the rs6983267 marker on chromosome 8. Cancer Res 68 9982 9986
21. YeagerM
OrrN
HayesRB
JacobsKB
KraftP
2007 Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet 39 645 649
22. GinzingerDG
GodfreyTE
NigroJ
MooreDH2nd
SuzukiS
2000 Measurement of DNA copy number at microsatellite loci using quantitative PCR analysis. Cancer Res 60 5405 5409
23. SnijdersAM
NoweeME
FridlyandJ
PiekJM
DorsmanJC
2003 Genome-wide-array-based comparative genomic hybridization reveals genetic homogeneity and frequent copy number increases encompassing CCNE1 in fallopian tube carcinoma. Oncogene 22 4281 4286
24. CurtinJA
FridlyandJ
KageshitaT
PatelHN
BusamKJ
2005 Distinct sets of genetic alterations in melanoma. N Engl J Med 353 2135 2147
25. R Development Core Team 2005 R: A Language and Environment for Statistical Computing Vienna, Austria R Foundation for Statistical Computing
26. GentlemanRC
CareyVJ
BatesDM
BolstadB
DettlingM
2004 Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol 5 R80
27. GajduskovaP
SnijdersAM
KwekS
RoydasguptaR
FridlyandJ
2007 Genome position and gene amplification. Genome Biol 8 R120
28. OlshenAB
VenkatramanES
LucitoR
WiglerM
2004 Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5 557 572
29. WillenbrockH
FridlyandJ
2005 A comparison study: applying segmentation to array CGH data for downstream analyses. Bioinformatics 21 4084 4091
30. BrownMB
ForsytheAB
1974 Robust tests for equality of variances. J Am Stat Assoc 69 364 367
31. WangC
HoriuchiA
ImaiT
OhiraS
ItohK
2004 Expression of BRCA1 protein in benign, borderline, and malignant epithelial ovarian neoplasms and its relationship to methylation and allelic loss of the BRCA1 gene. J Pathol 202 215 223
32. WeberF
ShenL
FukinoK
PatocsA
MutterGL
2006 Total-genome analysis of BRCA1/2-related invasive carcinomas of the breast identifies tumor stroma as potential landscaper for neoplastic initiation. Am J Hum Genet 78 961 972
33. van den BoomD
JurinkeC
McGinnissMJ
BerkenkampS
2001 Microsatellites: perspectives and potentials of mass spectrometric analysis. Expert Rev Mol Diagn 1 383 393
34. HaimanCA
Le MarchandL
YamamatoJ
StramDO
ShengX
2007 A common genetic risk factor for colorectal and prostate cancer. Nat Genet 39 954 956
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 9
Nejčtenější v tomto čísle
- Synthesizing and Salvaging NAD: Lessons Learned from
- Optimal Strategy for Competence Differentiation in Bacteria
- Long- and Short-Term Selective Forces on Malaria Parasite Genomes
- Identifying Signatures of Natural Selection in Tibetan and Andean Populations Using Dense Genome Scan Data