
Prions—Not Your Immunologist’s Pathogen
article has not abstract
Published in the journal:
. PLoS Pathog 11(2): e32767. doi:10.1371/journal.ppat.1004624
Category:
Pearls
doi:
https://doi.org/10.1371/journal.ppat.1004624
Summary
article has not abstract
A colleague and fellow immunologist, we will call her “Anne,” lifts her index and middle fingers on each hand and bows them in “air quotes” as she says prion “immunology” during my student’s thesis committee meeting. Anne says she works on “malaria, a real pathogen that elicits a real immune response.” Now, I am pretty sure Anne believes prions exist, but does she have a point about the immune response they elicit? The answer may surprise you.
Prions are remarkable, enigmatic pathogens that are quite different than most disease-causing entities. According to the prion hypothesis, prions are infectious agents devoid of instructional nucleic acid [1]. They propagate themselves without a genetic code, instead enciphering their infectious nature structurally, within the protein conformation itself. Mounting evidence supports the prion hypothesis, including the generation of infectious prions from purified recombinant protein [2]. Soon after Prusiner coined the term “prion,” his and Charles Weissmann’s labs discovered that a cellular gene encodes the prion agent [3]. Strangely, though, Prusiner had already demonstrated that infectious prions did not include nucleic acid, suggesting that prions infect without transmitting the gene encoding them. So attention turned to the host, in which this gene also encodes a normal form of the agent, called cellular prion protein (PrPC), that was later shown to be absolutely required to generate both genetic and acquired prion diseases [4]. And so, all the armchair immunologists reading this article right now pause and say, “Wait a minute…” while Anne chimes in with “prion immunology.” Here we go.
Current Evidence for an Immune Response to Prions
Natural prion exposure most likely involves the oronasal cavity and gastrointestinal tracts, both of which rely heavily on the immune system for protection from pathogens. Prion immunologists (no snickering, Anne) have put forth tremendous effort to characterize the host—prion interaction during infectious prion disease. Strong evidence demonstrates a significant role of innate immunity in both combatting and abetting peripheral prion pathogenesis [5]. Specialized epithelial cells of the mucosal immune system called microfold (M) cells sample luminal contents and pass them to innate immune cells residing on the other side in the lamina propria. M cells can transport prions from their apical surface contacting the lumen to their basolateral side to antigen presenting cells (APCs) waiting in the lamina propria.
APCs of the mononuclear phagocyte lineage, including macrophages, monocytes, and dendritic cells (DCs), process and present the majority of antigens introduced to the host immune system. All of these cells trap prions, with macrophages comprising most of them at the inoculation site [6]. These hungry cells remain there, gobbling up and sequestering, if not fully digesting, prions and preventing them from traveling to draining lymph nodes [7]. Those prions that escape macrophagocytosis can drain into and flow through lymph fluid (the immune system’s superhighway), unattached to cells, where they are trapped by another set of macrophages minding entry into the lymph node at the subcapsullary sinus. DCs and monocytes can also escort prions into draining lymph nodes later via innate immune molecules of the Complement system, including C1q, C3, and C4. DCs and monocytes express receptors for these Complement proteins that bind prion-Complement complexes. Toll-like receptors (TLRs), another class of innate immune receptors, may also aid this process [8]. B cells express Complement receptors CD21/35 that may bind prions directly without C3, which is required to mediate CD21/35 binding to conventional bacterial pathogens [9]. Even mast cells, the histamine cannons that mediate allergic immune responses, have been proposed to facilitate prion infection because they express significant PrPC and can be activated to release intracellular contents by binding C3 [5].
What happens next remains a mystery, but we do know that B cells, then another special antigen-presenting cell permanently residing in lymph nodes called follicular dendritic cells (FDCs), trap prions. B cells likely traffic prions to FDCs, which replicate them in nascent germinal centers forming in lymphoid follicles [8]. FDCs express loads of PrPC and moving FDCs closer to peripheral nerves increases the speed of prion neuroinvasion [10], while deleting them largely prevents disease [11]. Several studies have also reported changes in mRNA and protein levels of cytokines and chemokines, messengers and directors of immune responses, during prion infection.
Why No Adaptive Immune Response to Prions?
Anne rolls her eyes and says, “Yes, but what about a real immune response, one that adapts and responds to an infection with specificity and memory?” Well, that probably just does not happen. No humoral immune response to prions has been detected since researchers began looking in the early 1970s [12]. Presumably, negative selection eliminates B and T cells that recognize prions because they autoreact against PrPC, a self protein that shares identical primary amino acid sequence with PrPSc, the misfolded, prion form [13]. While this explanation comforts supporters of the prion hypothesis, immunologists like Anne remain skeptical. One might expect an adaptive immune response against prions, since one of their defining characteristics is misfolding the normal PrPC into PrPSc. This misfolding, or re-folding, should create secondary and tertiary structures that could be recognized by antibodies as novel, discontinuous epitopes that would be seen as foreign. A big problem, though, lies in another defining feature of prions—their resistance to protease digestion.
Explaining the Perverse and Often Baffling Lack of Prion-Specific Immunity
The binding of a protein to an antibody on the surface of a B cell is the first step toward the development of an antibody-producing plasma cell (Fig 1). B cells respond to this activation signal by endocytosing and degrading the protein into linear peptides. Class II MHC proteins display these peptides on the surface of B cells to T cells with a receptor specific for the peptide/MHC II complex. This interaction facilitates further signals including CD40 ligand binding to CD40 to help activate B cells to differentiate into plasma cells. Thus, optimal antibody production requires that peptide antigen be processed and presented on class II MHC, and T cells exist that recognize this peptide.
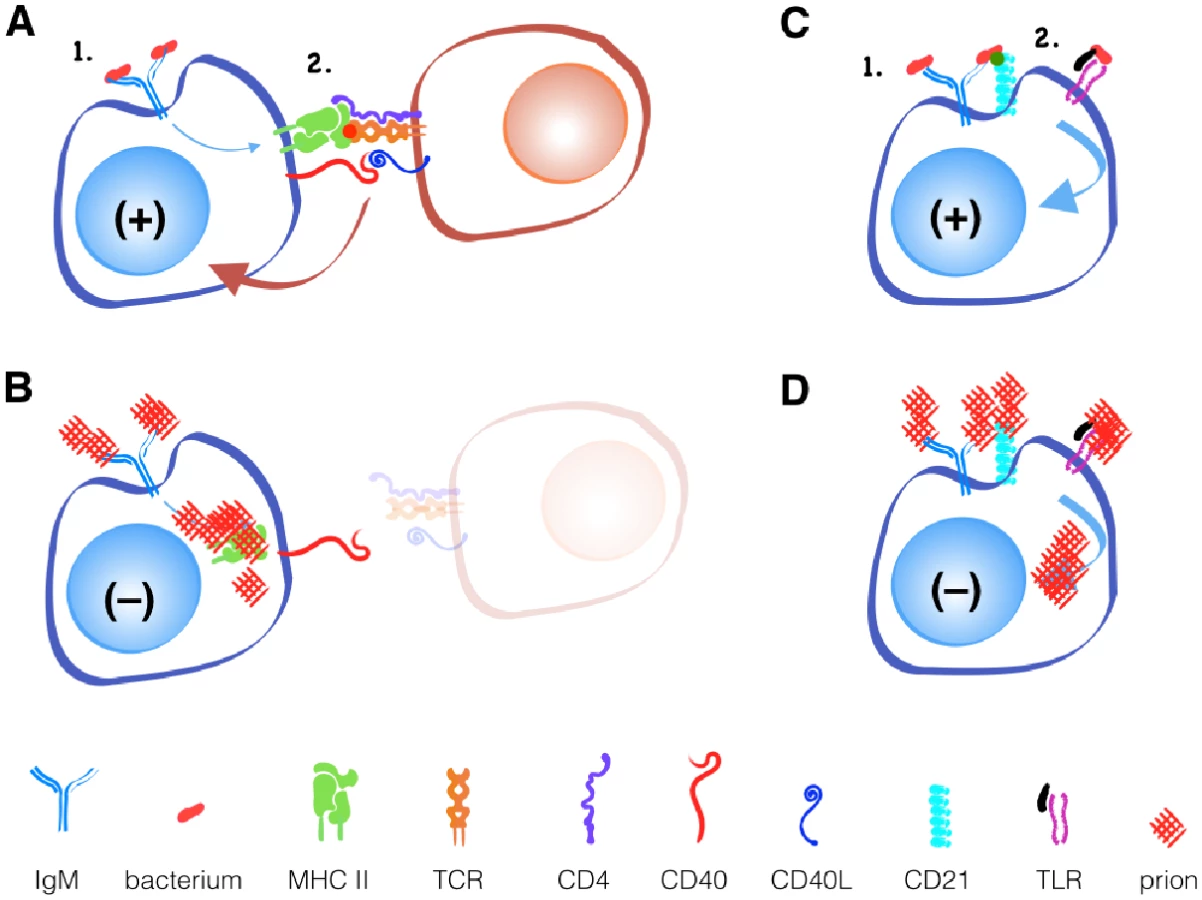
While B cell receptors may recognize novel discontinuous epitopes generated by PrPC misfolding to PrPSc, T cells recognize only linear epitopes. Since the primary amino acid sequence of PrPSc and PrPc is identical, T cells that recognize linear PrPSc will be recognized as self-reactive and will either be deleted in the thymus or controlled in the periphery via active inhibition by regulatory T cells or anergy via lack of inflammatory or other secondary stimuli.
An alternative, T cell-independent B cell activation can occur with polymeric antigens, typically non-protein antigens like bacterial polysaccharides and nucleic acids. These antigens can be recognized by a surface-bound antibody like IgM and other B cell receptors like CD21/35 and TLRs. Both receptors can provide secondary signals that help activate B cells without T cell help. If CD21/35 and TLRs can recognize prions, Anne argues, they should provide the secondary signal for B cell activation and antibody production that T cells cannot. So why does this not happen?
APCs, including B cells, typically process large extracellular antigens like aggregated prions via the external endosomal pathway (Fig 2). Endosomes internalize antigen then fuse with lysosomes containing acidic proteases like Cathepsins and aspargine endopeptidases that digest antigens into peptides that MHC II proteins present on the cell surface. Unfortunately, while prion infected cells increase expression of Cathepsins [14], these proteases may not effectively digest prions [15,16]. Accumulating prions may impair APCs’ ability to efficiently degrade cellular components in a process called autophagy [17], further limiting antigen processing and other cellular processes that keep APCs alive and kicking.
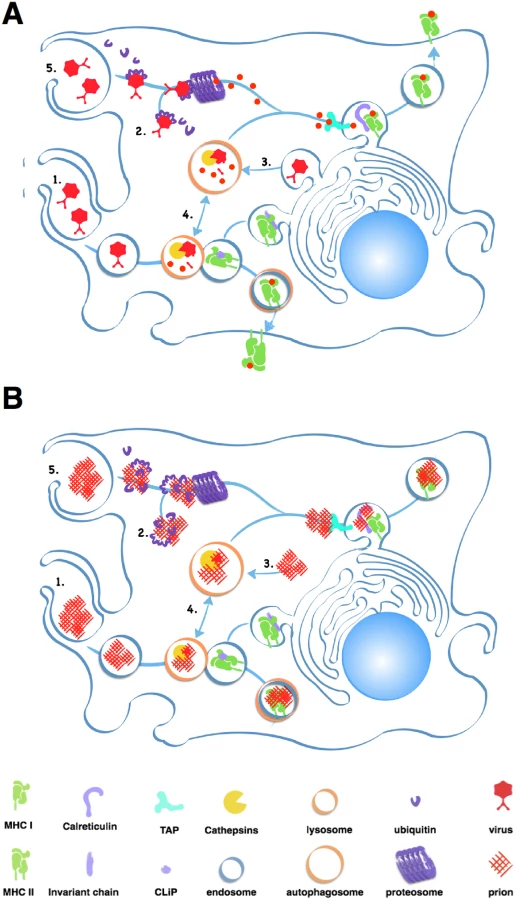
Prion-derived peptides may also have trouble gaining access to the class I presentation pathway. Cross-presentation of exogenous antigens like prions by class I MHC proteins can occur through the proteosome, a large protein-degrading complex that degrades cytosolic antigens and self protein tagged for disposal by chains of a small protein called ubiquitin. This endogenous pathway processes both self and foreign peptides for presentation on MHC class I molecules expressed on all nucleated cells. The proteosome typically processes smaller, soluble antigens, and prions inactivate this protein digester by directly inhibiting its proteolytic domain [18].
APCs may encounter further problems presenting prion peptides on MHC I proteins. Calreticulin, a protein important for loading peptide antigens into MHC I molecules, also binds C1q, a Complement protein that binds prions [19], which would likely block antigen loading. C3 can opsonize prions and APCs can phagocytose C3-opsonized immune complexes, but without antibodies, C3-coated prions enter endosomes that do not efficiently fuse with lysosomes, preventing further processing [20].
All told, prions present unique problems for both antibody production and CD8 T cell generation. B cells that recognize novel prion epitopes created by protein misfolding fail to receive sufficient secondary stimulation to differentiate into antibody-producing plasma cells. Prions constipate and stress APCs, causing lack of antigen processing and presentation and secondary signals that may explain why even prion protein deficient mice do not elicit antibody or cellular response to the misfolded protein.
Breaking Tolerance—Creating a Prion Vaccine
OK, so it seems that the adaptive immune system may not be able to process and/or present novel prion antigens. So what if we provide a pre-processed prion antigen, something that mimics a prion-specific epitope that APCs can present? Some success has been reported using aggregated or fragments of recombinant PrP [8,21]. Salmonella-expressing PrP stimulating mucosal immunity has been most successful so far [22]. Perhaps expressing a small, fibrillized prion peptide; prionoid amyloid; or other fibril mimetic would improve vaccine responses. Novel adjuvants like TLR agonists that provide powerful secondary signals important for robust vaccine responses, linking a prion peptide to a carrier protein like keyhole limpet hemocyanin that Cathepsins can easily cleave, or inflammatory monocyte migration inhibitors might further potentiate vaccine efficacy. Using soluble CD40 ligand as an adjuvant to bypass the requirement for T cell help may potentiate both humoral and cellular immune responses.
Be Careful What You Wish For—The Perverse and Often Baffling Immune Response to Prions
The holy grail of an effective vaccine is sterilizing immunity mediated by powerful neutralizing antibodies. Proof-of-principle has been shown in studies promoting mucosal immunity against prions [22]. The Complement system opsonizes most of the resulting antibody-antigen complexes, marking them for disposal mainly by Kupfer cells, the macrophages of the liver. However, compelling evidence shows that Complement facilitates prion transport to germinal centers within FDCs, where efficient prion replication occurs [9,23,24]. Generating prion-specific antibodies could therefore facilitate Complement trapping and transport of prions to draining lymph nodes—the very place they replicate most efficiently.
So maybe a cell-mediated response is better. Perhaps a more effective prion vaccine stimulates prion-specific T cells in the periphery to produce inflammatory cytokines like IFNγ, that can activate macrophages to phagocytose prions and degrade or at least sequester them, as has been shown to occur [6,7]. Such an atypical, cell-induced innate immune vaccine may be just what a host needs to respond to such an atypical pathogen. Or not, says Anne.
Zdroje
1. Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216: 136–144. 6801762
2. Wang F, Wang X, Yuan CG, Ma J (2010) Generating a Prion with Bacterially Expressed Recombinant Prion Protein. Science 327: 1132–1135. doi: 10.1126/science.1183748 20110469
3. Oesch B, Westaway D, Wälchli M, McKinley MP, Kent SBH, et al. (1985) A cellular gene encodes scrapie PrP 27–30 protein. Cell 40: 735–746. 2859120
4. Büeler HR, Aguzzi A, Sailer A, Greiner RA, Autenried P, et al. (1993) Mice devoid of PrP are resistant to scrapie. Cell 73: 1339–1347. Available: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgidbfrom=pubmed&id=8100741&retmode=ref&cmd=prlinks. 8100741
5. Bradford B, Mabbott N (2012) Prion Disease and the Innate Immune System. Viruses 4: 3389–3419. 23342365
6. Michel B, Meyerett-Reid C, Johnson T, Ferguson A, Wyckoff C, et al. (2012) Incunabular Immunological Events in Prion Trafficking. Sci Rep 2: 440. doi: 10.1038/srep00440 22679554
7. Beringue V, Demoy M, Lasmezas CI, Gouritin B, Weingarten C, et al. (2000) Role of spleen macrophages in the clearance of scrapie agent early in pathogenesis. J Pathol 190: 495–502. 10700001
8. Aguzzi A, Nuvolone M, Zhu C (2013) The immunobiology of prion diseases. Nat Rev Immunol 13: 888–902. doi: 10.1038/nri3553 24189576
9. Zabel MD, Heikenwalder M, Prinz M, Arrighi I, Schwarz P, et al. (2007) Stromal complement receptor CD21/35 facilitates lymphoid prion colonization and pathogenesis. J Immunol 179: 6144–6152. 17947689
10. Prinz M, Heikenwalder M, Junt T, Schwarz P, Glatzel M, et al. (2003) Positioning of follicular dendritic cells within the spleen controls prion neuroinvasion. Nature 425: 957–962. 14562059
11. Montrasio F, Frigg R, Glatzel M, Klein MA, Mackay F, et al. (2000) Impaired prion replication in spleens of mice lacking functional follicular dendritic cells. Science 288: 1257–1259. 10818004
12. Porter DD, Porter HG, Cox NA (1973) Failure to demonstrate a humoral immune response to scrapie infection in mice. J Immunol 111: 1407–1410. 4200779
13. Grégoire S, Bergot AS, Féraudet C, Carnaud C, Aucouturier P, et al. (2005) The murine B cell repertoire is severely selected against endogenous cellular prion protein. J Immunol 175: 6443–6449. 16272297
14. Zhang Y, Spiess E, Groschup MH, Burkle A (2003) Up-regulation of cathepsin B and cathepsin L activities in scrapie-infected mouse Neuro2a cells. Journal of General Virology 84: 2279–2283. Available: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12867662. 12867662
15. Luhr KM, Nordström EK, Löw P, Kristensson K (2004) Cathepsin B and L are involved in degradation of prions in GT-1 neuronal cells. Neuroreport 15: 1663–1667. Available: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15232303. 15232303
16. Yadavalli R, Guttmann RP, Seward T, Centers AP, Williamson RA, et al. (2004) Calpain-dependent endoproteolytic cleavage of PrPSc modulates scrapie prion propagation. J Biol Chem. 15764607
17. Heiseke A, Aguib Y, Schätzl HM (2010) Autophagy, prion infection and their mutual interactions. Curr Issues Mol Biol 12: 87–97. 19767652
18. Kristiansen M, Deriziotis P, Dimcheff DE, Jackson GS, Ovaa H, et al. (2007) Disease-Associated Prion Protein Oligomers Inhibit the 26S Proteasome. Molecular Cell 26: 175–188. 17466621
19. Sim RB, Kishore U, Villiers CL, Marche PN, Mitchell DA (2007) C1q binding and complement activation by prions and amyloids. Immunobiology 212: 355–362. 17544820
20. Kim SH, Visser A, Cruijsen C, van der Velden AWM, Boes M (2008) Recruitment of Rab27a to Phagosomes Controls Microbial Antigen Cross-Presentation by Dendritic Cells. Infection and Immunity 76: 5373–5380. doi: 10.1128/IAI.01044-08 18779337
21. Xanthopoulos K, Lagoudaki R, Kontana A, Kyratsous C, Panagiotidis C, et al. (2013) Immunization with recombinant prion protein leads to partial protection in a murine model of TSEs through a novel mechanism. PLoS ONE 8: e59143. doi: 10.1371/journal.pone.0059143 23554984
22. GONI F, Prelli F, SCHREIBER F, SCHOLTZOVA H, Chung E, et al. (2008) High titers of mucosal and systemic anti-PrP antibodies abrogate oral prion infection in mucosal-vaccinated mice. Neuroscience 153: 679–686. doi: 10.1016/j.neuroscience.2008.02.051 18407424
23. Mabbott NA, Bruce ME, Botto M, Walport MJ, Pepys MB (2001) Temporary depletion of complement component C3 or genetic deficiency of C1q significantly delays onset of scrapie. Nat Med 7: 485–487. Available: http://www.ncbi.nlm.nih.gov/cgi-bin/Entrez/referer?http://www.nature.com/cgi-taf/DynaPage.taf%3ffile=/nm/journal/v6/n7/full/nm0700_719.html. 11283677
24. Klein MA, Kaeser PS, Schwarz P, Weyd H, Xenarios I, et al. (2001) Complement facilitates early prion pathogenesis. Nat Med 7: 488–92. 11283678
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 2
- Měli bychom postcovidový syndrom léčit antidepresivy?
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Control of Murine Cytomegalovirus Infection by γδ T Cells
- ATPaseTb2, a Unique Membrane-bound FoF1-ATPase Component, Is Essential in Bloodstream and Dyskinetoplastic Trypanosomes
- Rational Development of an Attenuated Recombinant Cyprinid Herpesvirus 3 Vaccine Using Prokaryotic Mutagenesis and In Vivo Bioluminescent Imaging
- Telomeric ORFS in : Does Mediator Tail Wag the Yeast?