
The Pneumococcal Serine-Rich Repeat Protein Is an Intra-Species Bacterial Adhesin That Promotes Bacterial Aggregation and in Biofilms
The Pneumococcal serine-rich repeat protein (PsrP) is a pathogenicity island encoded adhesin that has been positively correlated with the ability of Streptococcus pneumoniae to cause invasive disease. Previous studies have shown that PsrP mediates bacterial attachment to Keratin 10 (K10) on the surface of lung cells through amino acids 273–341 located in the Basic Region (BR) domain. In this study we determined that the BR domain of PsrP also mediates an intra-species interaction that promotes the formation of large bacterial aggregates in the nasopharynx and lungs of infected mice as well as in continuous flow-through models of mature biofilms. Using numerous methods, including complementation of mutants with BR domain deficient constructs, fluorescent microscopy with Cy3-labeled recombinant (r)BR, Far Western blotting of bacterial lysates, co-immunoprecipitation with rBR, and growth of biofilms in the presence of antibodies and competitive peptides, we determined that the BR domain, in particular amino acids 122–166 of PsrP, promoted bacterial aggregation and that antibodies against the BR domain were neutralizing. Using similar methodologies, we also determined that SraP and GspB, the Serine-rich repeat proteins (SRRPs) of Staphylococcus aureus and Streptococcus gordonii, respectively, also promoted bacterial aggregation and that their Non-repeat domains bound to their respective SRRPs. This is the first report to show the presence of biofilm-like structures in the lungs of animals infected with S. pneumoniae and show that SRRPs have dual roles as host and bacterial adhesins. These studies suggest that recombinant Non-repeat domains of SRRPs (i.e. BR for S. pneumoniae) may be useful as vaccine antigens to protect against Gram-positive bacteria that cause infection.
Published in the journal:
. PLoS Pathog 6(8): e32767. doi:10.1371/journal.ppat.1001044
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001044
Summary
The Pneumococcal serine-rich repeat protein (PsrP) is a pathogenicity island encoded adhesin that has been positively correlated with the ability of Streptococcus pneumoniae to cause invasive disease. Previous studies have shown that PsrP mediates bacterial attachment to Keratin 10 (K10) on the surface of lung cells through amino acids 273–341 located in the Basic Region (BR) domain. In this study we determined that the BR domain of PsrP also mediates an intra-species interaction that promotes the formation of large bacterial aggregates in the nasopharynx and lungs of infected mice as well as in continuous flow-through models of mature biofilms. Using numerous methods, including complementation of mutants with BR domain deficient constructs, fluorescent microscopy with Cy3-labeled recombinant (r)BR, Far Western blotting of bacterial lysates, co-immunoprecipitation with rBR, and growth of biofilms in the presence of antibodies and competitive peptides, we determined that the BR domain, in particular amino acids 122–166 of PsrP, promoted bacterial aggregation and that antibodies against the BR domain were neutralizing. Using similar methodologies, we also determined that SraP and GspB, the Serine-rich repeat proteins (SRRPs) of Staphylococcus aureus and Streptococcus gordonii, respectively, also promoted bacterial aggregation and that their Non-repeat domains bound to their respective SRRPs. This is the first report to show the presence of biofilm-like structures in the lungs of animals infected with S. pneumoniae and show that SRRPs have dual roles as host and bacterial adhesins. These studies suggest that recombinant Non-repeat domains of SRRPs (i.e. BR for S. pneumoniae) may be useful as vaccine antigens to protect against Gram-positive bacteria that cause infection.
Introduction
Streptococcus pneumoniae is a leading cause of otitis media (OM), community-acquired pneumonia, sepsis and meningitis. Primarily a commensal, S. pneumoniae typically colonizes the nasopharynx asymptomatically, however in susceptible individuals such as infants, the elderly, persons who are immunocompromised, and those with sickle cell anemia, the pneumococcus is often able to cause opportunistic diseases [1], [2], [3], [4]. Worldwide, S. pneumoniae is responsible for up to 14.5 million episodes of invasive pneumococcal disease (IPD) and 11% of all deaths in children [5], [6]. In the elderly the mortality-rate associated with IPD can exceed 20% and for those in nursing homes may be as high as 40% [7]. Thus, the pneumococcus has been and remains a major cause of morbidity and mortality.
psrP-secY2A2 is a S. pneumoniae pathogenicity island whose presence has been positively correlated with the ability to cause human disease [8]. Analyses of the published S. pneumoniae genomes has demonstrated that psrP-secY2A2 is present and conserved in a number of globally distributed invasive clones, in particular those belonging to serotypes not covered by the heptavalent conjugate vaccine [9]. To date, numerous studies have shown that deletion of genes within psrP-secY2A2 attenuated the ability of S. pneumoniae to cause disease in mice. psrP-secY2A2 mutants were shown to be unable to attach to lung cells, establish lower respiratory tract infection, and were delayed in their ability to enter the bloodstream from the lungs. Importantly, the same studies found that psrP-secY2A2 did not play an important role during nasopharyngeal colonization or during sepsis following intraperitoneal challenge [10], [11], [12], [13]. Thus psrP-secY2A2 is currently understood to be a lung-specific virulence determinant.
In TIGR4, a virulent serotype 4 laboratory strain, psrP-secY2A2 is 37-kb in length and encodes 18 proteins. These include the Pneumococcal serine-rich repeat protein (PsrP), which is a lung cell adhesin, 10 putative glycosyltranferases, and 7 proteins homologous to components of an accessory Sec translocase [14]. To date, the latter 17 genes remain uncharacterized; however, based on their homology to genes found within the Serine-rich repeat protein (SRRP) locus of Streptococcus gordonii, the encoded proteins are putatively responsible for the intracellular glycosylation of PsrP and for its transport to the bacterial surface [8], [15], [16], [17], [18]. PsrP in TIGR4 is composed of 4,776 amino acids, has been confirmed to be glycosylated, and separates at an apparent molecular mass of 2,300 kDa on an agarose gel [13]. It is one of the largest bacterial proteins known. PsrP is organized into multiple domains including a cleavable N-terminal signal peptide, a small serine-rich repeat region (SRR1), a unique non-repeat region (NR), followed by a second extremely long serine-rich region (SRR2), and a C-terminal cell wall anchor domain containing an LPXTG motif (Figure 1A). The SRR1 and SRR2 domains of PsrP are composed of 8 and 539 serine-rich repeats (SRR) of the amino acid sequence SAS[A/E/V]SAS[T/I], respectively, and are the domains believed to be glycosylated. The NR domain of PsrP has a predicted pI value of 9.9, for this reason it is called the Basic Region (BR) domain.

S. pneumoniae is surrounded by a polysaccharide capsule that protects the bacteria from phagocytosis but also inhibits adhesion to epithelial cells [19]. Based on the size and domain organization of PsrP we have previously hypothesized that the extremely long SRR2 domain serves to extend the BR domain through the capsular polysaccharide to mediate lung cell adhesion (Figure 1B) [12], [13]. Consistent with this model, we have previously shown that PsrP is expressed on the bacterial surface, that the BR domain, in particular amino acids 273–341, was responsible for PsrP-mediated adhesion to Keratin 10 (K10) on lung cells, and that complementation of psrP deficient mutants with a truncated version of the protein (having only 33 SRRs in its SRR2 domain) restored the ability of uncapsulated but not capsulated PsrP mutants to adhere to A549 cells, a human type II pneumocyte cell line [13].
It is now recognized that biofilms play an important role during infectious diseases. Briefly, bacteria in biofilms are more resistant to host-defense mechanisms including phagocytosis and serve as a recalcitrant source of bacteria during antimicrobial therapy [20], [21]. For S. pneumoniae, pneumococcal biofilms have been shown to occur in the middle ears of children with chronic otitis media and is thought to contribute to its refractory nature [22]. Likewise, biofilms have been detected in the nasopharynx of infected chinchillas [23]. However, until now biofilm structures have not been described in the lungs during pneumococcal pneumonia. This is in contrast to other respiratory tract pathogens, such as Pseudomonas aeruginosa and Bordatella pertussis, for which in vivo biofilm production is now recognized to be an important pathogenic mechanism [21]. Herein, we demonstrate for the first time that S. pneumoniae forms biofilm-like aggregates in the lungs. We show that this phenomenon is PsrP-dependent and mediated by its BR domain. Using recombinant protein and SRRP mutants, we show that the SRRPs of S. gordonii and Staphylococcus aureus, GspB and SraP, respectively, also promote bacterial aggregation, thus describing a previously unrecognized role for members of the SRRP family. Collectively, these findings suggest an important dual role for PsrP and other SRRPs during infection, host cell and intra-species bacterial adhesion, both of which may be targeted for intervention with antibodies against recombinant (r)NR.
Results
PsrP promotes pneumococcal aggregation in vivo
To test whether PsrP contributed to biofilm or microcolony formation in vivo mice were infected with TIGR4 and its isogenic psrP deficient mutant, T4 ΔpsrP, and whole lung sections were examined using scanning electron microscopy (SEM). As would be expected for both wild type and the mutant, the majority of bacteria present were in the form of diplococci. However, for TIGR4 we also observed the presence of large bacterial aggregates attached to ciliated bronchial epithelial cells as well as to alveolar epithelial cells (Figure 2). For quantitative analysis of this phenomenon, nasal lavage fluid and bronchoalveolar lavage (BAL) fluid from mice was collected two days post-challenge. Aliquots from each biological sample were heat-fixed to glass slides, Gram-stained, and examined with a microscope (Figure 3A). In all, the number of bacterial aggregates composed of 2–9, and ≥10 diplococci were significantly greater for mice infected with TIGR4 than T4 ΔpsrP in both the nasopharyngeal and BAL elute fluids (Figure 3B,C). Moreover, the largest aggregates, those composed of >100 bacteria, were observed only in mice infected with TIGR4. Fluorescent imaging of bacteria in frozen lung sections confirmed this phenotype; large bacterial aggregates were only detected in the lungs of TIGR4 infected mice (Figure S1). Thus we determined that PsrP promoted the formation of biofilm-like aggregates in vivo, including in the nasopharynx, a site previously shown not to require PsrP for bacterial colonization [12].


PsrP affects intimate bacteria to bacteria interactions
Given the previous results, moreover to develop an in vitro model that was amendable to manipulation, the ability of TIGR4 and T4 ΔpsrP to form early biofilms was tested using microtiter plates [24]. As shown in Figure 4A, no differences were observed between wild type and the mutant, suggesting that PsrP does not play a role in pneumococcal attachment to polystyrene or the formation of early biofilm structures, in particular the bacteria lawn. The role of PsrP was next tested in 3-day old mature biofilms using the once-through continuous flow cells as described previously by Allegrucci et al. [25]. In this system, a stark difference in the architecture of TIGR4 and T4 ΔpsrP biofilms was observed (Figure 4B). Wild type biofilms displayed a dense cloud-like morphology with extremely large aggregates that covered the glass surface. Closer inspection revealed that these aggregates were composed of tightly clustered pneumococci. In contrast, T4 ΔpsrP biofilms displayed a less intimate phenotype characterized by smaller aggregates, gaps, and the formation of columns, resulting in an overall patchier phenotype. Quantitative analysis of the biofilm structures using COMSTAT software confirmed that TIGR4 biofilms had significantly greater total biomass and average thickness than those formed by the T4 ΔpsrP (Figure 4C). No differences in either the maximum thickness of the biofilms or the roughness coefficient (a measure of biofilm heterogeneity) were observed (Figure 4C; data not shown, respectively), indicating that T4 ΔpsrP could still form biofilms, although with distinct architecture. Importantly, T4 ΩpsrP-secY2A2, a mutant deficient in the entire psrP-secY2A2 pathogenicity island, behaved identically to T4 ΔpsrP, forming patchy biofilms with small aggregates and less intimate associated bacteria (Figure S2).

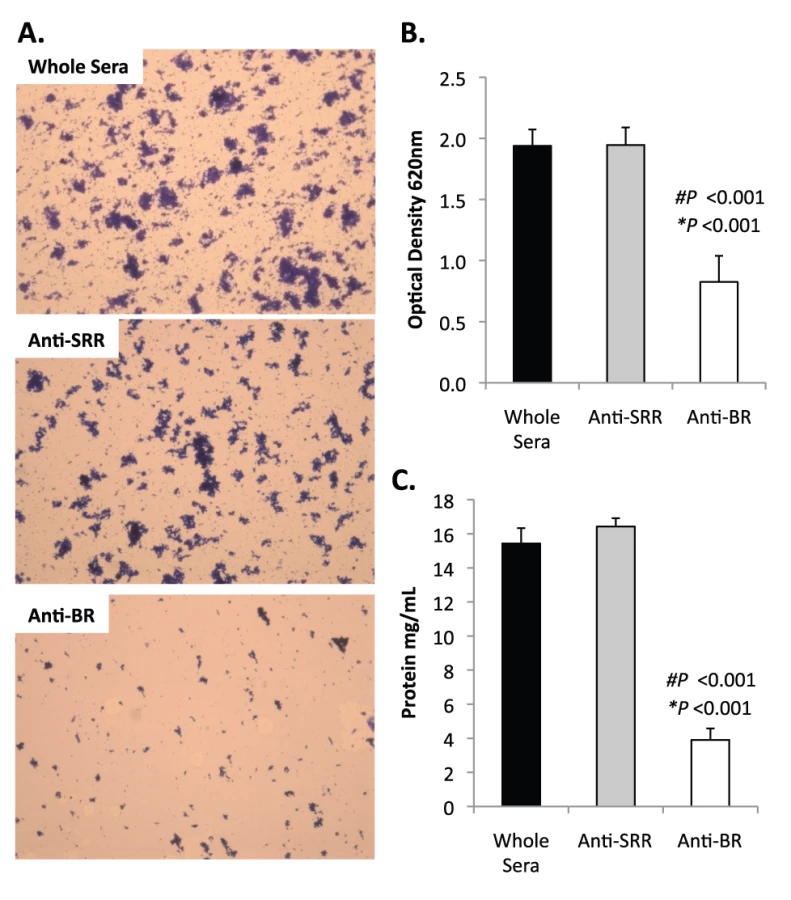
Bacterial biofilms were also grown under once through conditions in silicone tubing. After a designated time, the biofilms were extruded from the line and examined for biomass both visually and quantitatively. After 3 days of growth, differences between TIGR4 and T4 ΔpsrP in opacity of the exudates were visible to the eye (Figure 5A) and could be confirmed using a spectrophotometer which showed a >3-fold difference in optical density (Figure 5B). Microscopic visualization of the line exudates following crystal violet (CV) staining revealed that TIGR4 had formed large aggregates whereas T4 ΔpsrP exudates were composed of small clusters or of individual diplococci (Figure 5C). Increased biofilm biomass was supported by measurement of total protein concentrations that showed TIGR4 biofilm exudates had 2–3 fold more protein than those corresponding to T4 ΔpsrP (Figure 5D).

Of note, during planktonic growth TIGR4, T4 ΔpsrP, and T4 ΩpsrP-secy2A2 were indistinguishable, growing either as short chains or diplococci with a marked absence of aggregates (data not shown). This led us to examine psrP transcription using Real-Time PCR and the finding that TIGR4 expressed psrP at levels 47-fold greater during biofilm versus planktonic culture (P = 0.04 using a Student's t-test). Thus low expression of psrP may be one reason TIGR4 did not form aggregates during liquid culture.
The BR domain mediates intra-species bacterial interactions
To date a number of groups, including our own, have shown that SRRPs mediate bacterial adhesion to host cells primarily through their NR domain [13], [26], [27]. For this reason we sought to test whether the BR domain of PsrP was also involved in biofilm/bacterial aggregation. To do this we first utilized a pre-existing collection (described in Figure S3) of encapsulated (T4 ΩpsrP) and unencapsulated (T4R ΩpsrP) S. pneumoniae mutants deficient in PsrP that either expressed a truncated version of PsrP with 33 SRRs in its SRR2 domain (PsrPSRR2(33)), a similar truncated version lacking the BR domain (PsrPSRR2(33)-BR), or carried the empty expression vector pNE1 [13]. These strains were tested for their ability to form biofilms in silicone lines under once through conditions.
Complementation of T4 ΩpsrP with PsrPSRR2(33), but not PsrPSRR2(33)-BR or the empty pNE1 vector, partially restored the ability of T4 ΩpsrP to form large aggregates in the lines when examined microscopically (Figure 6A). However, measurement of other biofilm markers such as optical density and total protein concentration showed no differences between any of the complemented mutants and the negative controls (Figure 6B–C). Complementation of T4R ΩpsrP with PsrPSRR2(33), also partially restored the ability of T4R ΩpsrP to form aggregates (Figure 6A). In this instance, line exudates from T4R ΩpsrP with PsrPSRR2(33) had significant more biofilm biomass than the negative controls (Figure 6B–C). Importantly, the truncated version of PsrP lacking the BR domain failed to restore, even partially, T4 ΩpsrP or T4R ΩpsrP suggesting that the BR domain was responsible for the intra-species aggregation. This was subsequently confirmed by Far-Western blot analyses that showed that Gst-tagged recombinant BR (Gst-BR) bound only to S. pneumoniae cell lysates that contained a truncated PsrP with the BR domain (Figure 6D) and a control experiment showing that a Gst-tagged Chlamydia trachomatis protein did not interact with these lysates (Figure S4).

To further explore the role of the BR domain in the observed bacteria to bacteria interactions, the ability of His-tagged BR constructs (rBR; Figure 7A), purified from Escherichia coli and Cy3 labeled, were tested for their ability to bind to the surface of TIGR4 and T4 ΔpsrP. Full-length rBR interacted with TIGR4 but not with T4 ΔpsrP (Figure 7B), confirming not only that PsrP bound to pneumococci, but also suggesting that its ligand was another PsrP. Furthermore, only rBR.A retained the ability to attach to PsrP on the pneumococcal surface. This suggested that the binding domain of PsrP was possibly located within AA 122–166, the section not shared between rBR.A and rBR.B.

Hereafter, BR to BR interactions were tested for by Far Western and co-immunoprecipitation. Far Western blot experiments using assorted E. coli cell lysates from bacteria expressing assorted rBR constructs, confirmed that only lysates containing PsrP constructs with AA 122–166 bound successfully to Gst-BR (Figure 7C). This was also observed in co-immunoprecipitation experiments, whereby Gst-BR was tested for its ability to bind whole cell lysates from E. coli expressing versions of PsrP (Figure 7D). Far Western blots using purified proteins showed that Gst-BR had affinity to purified rBR, rBR.A, and a synthesized peptide corresponding to AA 122–166, but not rBR.B, BR.C, or the control his-tagged Streptolysin O (Figure 7E). Hence, using numerous assays it was determined that the BR domain, most likely AA 122–166, had self-interacting properties that might be responsible for the observed bacterial aggregation.
Of note, because the BR constructs were purified from E. coli and PsrP is normally glycosylated, the above observations may have been an artifact of the unglycosylated constructs used. To address this possibility a glycosyated truncated PsrP construct was purified from S. pneumoniae (PsrPSRR2(33)-HIS; Figure S5) and tested for its ability to bind S. pneumoniae cell lysates containing either native PsrP or assorted constructs. As shown in Figure 7F, it was determined that a glycosylated PsrP probe maintained specificity for the BR domain even in the context of glycosylated recipient protein. A finding that supports the notion that PsrP to PsrP interactions occur in natural setting when PsrP is always glycosylated.
The aggregation and K10-binding subdomains of BR are independent
To determine whether the BR aggregation (AA 122–167) and the K10 binding subdomains (AA 273–341) of BR had functionally independent roles, competitive inhibition assays were performed using rBR constructs. Bacterial adhesion to A549 cells was tested following incubation of cells with the AA 122–166 peptide, rBR, and rBR.C (Figure 8A). Pre-treatment of A549 cells with AA 122–167 had no impact on adhesion. In contrast and consistent with the location of the K10 binding domain within BR.C: 1) TIGR4 adhered significantly less to cells treated with rBR or rBR.C, 2) TIGR4 adhered to BSA treated cells better than T4 ΔpsrP. In complementary biofilm experiments the opposite result was observed. Addition of 1 µM peptide AA 122–167 to media reduced the aggregation phenotype observed for TIGR4 (Figure 8B) and modestly lowered the optical density of the biofilm exudate and the total biomass collected from the continuous flow lines versus addition of BR.C (Figure 8C–D). Thus these findings suggested that the aggregation and K10 subdomains of PsrP had distinct roles that did not overlap during host cell adhesion or biofilm formation.

Finally we sought to determine a biological effect for the aggregation phenotype. We observed that after 1 hour, 69±2% of J477 macrophages incubated with planktonically grown TIGR4 were associated with FITC-labeled bacteria whereas only 51±5% of macrophages mixed with biofilm grown TIGR4 were positive (P = 0.024). Macrophages exposed to biofilm grown TIGR4 also took up less bacteria than macrophages mixed with planktonic (74±1%; P = <0.001) and biofilm (60±1%; P = <0.001) cultures of T4 ΔpsrP. Interestingly, a 10% reduction in macrophage uptake was observed for the biofilm versus planktonic grown T4 ΔpsrP cultures (P = 0.077); and no difference was observed between macrophage uptake of TIGR4 and T4 ΔpsrP when taken from planktonic cultures. These findings suggest, that in addition to PsrP, other bacterial factors expressed during growth in a biofilm also affect opsonophagoyctosis.
Antibodies to the BR domain, but not to the SASASAST motif, block bacterial aggregation
Previously we had shown that antibodies against the SRR1-BR domains of PsrP neutralized the ability of S. pneumoniae to attach to lung cells and that vaccination with rBR protected mice against pneumococcal challenge [12], [13]. For this reason we tested the ability of polyclonal antiserum against rBR and against a SRR motif peptide to block bacterial aggregation in the biofilm line model. Todd Hewitt Broth (THB) supplemented with a 1∶1000 dilution of antiserum against the BR domain inhibited the formation of bacterial aggregates as observed by microscopic visualization of the biofilm line exudates. In contrast, bacteria in media supplemented with antiserum to the SRR motif peptide or that from naïve animals, formed aggregates similar to wild type bacteria grown under serum free conditions (Figure 9A). Biofilm exudate optical density and protein concentrations supported these microscopic observations (Figure 9B–C). To determine whether the effect of the BR antiserum on biofilm formation was specific for TIGR4, we tested the ability of antibodies to the BR domain to block biofilm formation in unrelated clinical isolates (Figure S6). Antiserum against rBR from TIGR4 inhibited biofilm formation in two unrelated clinical isolates that carried PsrP. The same sera had no effect on biofilm formation by an invasive serotype 14 isolate that lacked PsrP. Therefore these studies confirmed previous observations that increased bacteria aggregation in biofilm models can occur independently of PsrP, but that if present, antiserum against BR can block the contribution of PsrP to these processes.
SRRPs mediate intra-species adhesion in pathogenic bacteria
To determine whether other SRRPs also mediated intra-species aggregation we tested the effect of gspB and sraP deletion on S. gordonii and S. aureus biofilm architecture, respectively. Deletion of gspB and sraP negatively impacted biofilm formation in the microtiter biofilm model at 24 hours (Figure 10A,B). Growth of wild type and mutant bacteria in the line models also demonstrated that both proteins contributed to the formation of large aggregates during surface attached growth; although this property was much more dramatic for S. gordonii than for S. aureus which did not show a significant difference in the optical densities of the exudates (Figure 10C,D). Of note, S. aureus biofilm experiments were stopped after 1 day due to bacteria overgrowth and blockage of the lines.

Subsequent Far Western analysis using Gst-BR from S. pneumoniae as well as recombinant SRR1-NR from SraP and recombinant NR from GspB showed that these proteins have affinity for cell lysates from their parent strain but not for cell lysates from isogenic SRRP deficient mutants (Figure 10E). This supports the notion that these proteins might be involved in intra-species aggregation. For PsrP BR from S. pneumoniae, no affinity was observed for cell lysates from either S. gordonii or S. aureus suggesting that PsrP does not play a role as an inter-species adhesin (Figure 10E). In contrast, the NR constructs from S. aureus and S. gordonii bound to cell lysates from the other bacteria, even in the absence of the SRRP (Figure 10E). The discrepancy between PsrP and the other SRRPs might be explained by the fact that certain SRRPs have been described to have lectin activity [26], [27]. In contrast PsrP adhesion has been shown to be independent of lectin-activity [13].
Discussion
To date, SRRPs have been described in at least 9 Gram-positive bacteria and have been shown to function as adhesins that contribute to virulence. For example, deletion of sraP and gspB in S. aureus and S. gordonii, respectively, decreased the ability of these bacteria to bind to platelets and form vegetative plaques on heart valves of catheterized rats [27], [28]. Similarly, Srr-1 of Streptococcus agalactiae has been shown to bind human Keratin 4, mediate adherence to mucosal epithelial cells, and promote invasion of bacteria through human brain microvasculature endothelial cells [29], [30]. SRRPs also mediate acellular attachment, a role important for colonization of the dental surface by oral streptococci. Froelinger and Fives-Taylor showed that Streptococcus parasanguis containing mutations of Fap1 failed to attach to saliva-coated hydroxyapatite [31]. Likewise, deletion of srpA significantly diminished the ability of Streptococcus cristatus to attach to glass slides [32]. Thus, while it was well established that SRRPs play an important role in bacterial attachment to cells or surfaces, until this report their role as intra-species adhesins remained unrecognized.
A dual role, host and bacterial adhesin for bacterial surface proteins is not unprecedented. For example, in Streptococcus pyogenes and S. agalactiae, the pilus proteins mediate adhesion to epithelial cells and promote microtiter biofilm formation [33], [34]. Likewise, for Neisseria meningitidis, PilX, also a pilus-associated protein, mediates adhesion to epithelial cells and facilitates bacterial aggregation [35]. For the pneumococcus, some evidence existed that bacterial adhesins may also have dual roles. In 2008, Munoz-Elias et al. found that the pneumococcal adhesins Choline binding protein A and the pilus protein RrgA were both required for robust biofilm formation on microtiter plates and efficient nasopharyngeal colonization [36]. However, the attenuated biofilm phenotype was observed only with unencapsulated bacteria and encapsulated mutants formed biofilms normally. Other pneumococcal proteins shown to affect biofilm formation in vitro include Neuraminidase A, which possibly alters the extracellular matrix [37], [38], [39], competence proteins, which suggest an altered protein profile [40], [41], and capsule synthesis enzymes, which were determined to be down regulated in biofilms [36], [42], [43]. Unlike PsrP, which would be expected to bridge cells directly, these proteins most likely act indirectly by altering gene expression, the extracellular milieu, or the surface availability of other adhesins, including possibly RrgA and CbpA.
Our studies determined that the self-aggregating subdomain of PsrP was located in the BR domain and involves amino acids 122–166. Recombinant BR constructs containing these amino acids were able to bind S. pneumoniae carrying PsrP, had an affinity for the BR domain in other PsrP constructs, and could modestly inhibit biofilm formation when added to media. Importantly, adhesion assays using pretreated cells and biofilm assays with rBR.C showed that the AA 122–166 was not responsible for adhesion to lung cells and that the K10 binding subdomain (AA 273–341) was not involved in bacterial aggregation. Thus these subdomains appeared to have independent roles during the conditions tested. Further studies are warranted to delineate the specific AAs responsible for these adhesive properties, also to determine the structure of the BR domain and clarify how these subdomains interact with PsrP on other pneumococci and K10 on lung cells.
GspB and SraP have been previously shown to bind platelets [27], [28]. While the ligand for SraP is unknown, it has been determined that GspB binds to Sialyl T-antigen on platelet membrane glycoprotein Ibα [26], [27]. The observation that the NR domains of GspB and SraP bound to cell lysates containing their respective SRRPs but not to their mutants and that the mutants had diminished aggregative properties suggests that SRRPs in other bacteria might also mediate aggregation in vivo. One could imagine that SraP on S. aureus or GspB on S. gordonii mediating attachment to platelets and cells in an endocarditic lesion while at the same time mediating adhesion of individual bacteria to each other. Similarly, one could envision a microcolony of the pneumococcus in the lungs with some bacteria attached to host cells via PsrP/K10 interactions and other bacteria attached to these bacteria through PsrP/PsrP interactions. Presumably, this is what was observed in the lungs of the infected mice. Interestingly, the finding that GspB and SraP NRs bound to cell lysates from other bacteria suggests that these proteins may also mediate inter-species biofilm formation. For S. gordonii, this would be relevant as the dental plaque is now recognized to be a multi-species biofilm. Importantly, neutralization of pneumococcal aggregation in biofilms with BR antiserum suggests that SRRPs might have utility as vaccine antigens. One caveat is that SRRPs would have to be one-component of a multi-valent vaccine because not all strains of S. pneumoniae, S. aureus, or the oral streptococci carry these proteins.
In previous studies we had found that the length of the SRR2 domain was important for adhesion to K10 when capsule was present. Consistent with these findings, the inability of truncated PsrP to fully complement capsulated mutants supports our hypothetical model that the SRR2 domain serves to extend the BR domain away from the cell to mediate bacterial interactions. This model is also indirectly supported by Munoz-Elias et al., who showed that down-regulation of capsule allowed CbpA and RrgA to contribute to biofilm production [36]. It is also noteworthy to state that Munoz-Elias et al. did not identify PsrP in their screen for biofilm mutants although they used TIGR4 which carries PsrP. This can be explained by the fact that we observed no contribution for PsrP in the microtiter plate early biofilm model.
We observed that PsrP-mediated bacterial aggregation occurred in the nasopharynx, despite earlier studies demonstrating that K10 was absent from this site and that PsrP was not required for nasopharyngeal colonization. Aggregation of S. pneumoniae in the nasopharynx may serve as a mechanism to resist opsonophagocytosis as shown herein, or we speculate a way to resist desiccation during transmission of infectious particles. The observation that aggregates were present at an anatomical site that lacked K10, further supports an independent role for these PsrP subdomains. In regards to opsonophagoyctosis, one important consideration is that the pneumococcus most likely has different gene expression profiles in vivo as an aggregate attached to a cell versus in vitro as a biofilm [44]. Thus caution is warranted in applying our vitro observations, such as resistance to opsonophagoyctosis or enhanced PsrP expression during biofilm growth, with events that occur in vivo.
Polyclonal antibodies against the BR domain, but not the SRR motif, neutralized the ability of TIGR4 and clinical isolates carrying PsrP to form aggregates in the line model. These findings were consistent with previous studies showing that antibodies against BR also neutralized its ability to mediate adhesion to host cells and protected mice against pneumonia [12], [13]. One possible reason that antibodies against the SRR motif peptide failed to have a neutralizing effect is that PsrP is glycosylated and antibodies against the peptide failed to recognize the native version of the protein. Alternatively, antibodies to the SRR motif may bind away from the BR domain and therefore do not inhibit the ability of the BR domain to self-interact. Interestingly, polyclonal antibodies to surface proteins often promote aggregation. This did not occur for unknown reasons. Finally, our finding that antibodies against rBR neutralized bacterial aggregation in the biofilm line model suggests that the same antibodies might also neutralize bacterial aggregation in vivo. This remains to be tested, however, the protection that was observed in mice following immunization with rBR [13], may have been in part due to inhibition of bacterial aggregation in addition to blocking interactions with K10.
Importantly, because rNR domains produced in E. coli are not glycosylated, yet for the tested SRRPs were able to aggregate, immunoprecipitate, and bind to native protein in cell lysates, it seems that the BR domain does not require glycosylation to function as a self-adhesin. This is supported by the observation that addition of antibodies against unglycosylated rBR and that synthetic peptide AA 122–166 both inhibited bacterial aggregation in the biofilm line. In contrast to the latter concept, Wu et al. demonstrated that monoclonal antibodies specific for the glycan motifs of the serine-rich repeat motifs of Fap1 were capable of blocking attachment to saliva coated hydroxyapatite by Streptococcus parasanguis [45]. Importantly, Fap1 is the most divergent of the SRRPs and has 2 NR domains. Fap1 adhesion to saliva coated hydroxyapatite is mediated by glyconjugates on the serine-rich repeat domain [46]; as evidenced by the fact that inactivation of one of the glycosyltranferases known to modify the glycan moieties of Fap1, drastically altered the ability of S. parasanguis to form biofilms [45]. Thus Fap1 is interesting because it suggests an NR-independent mechanism for SRRP adhesion, which is distinct from those discussed for GspB, SraP, or PsrP. Future studies need to further examine the differences between these diverse SRRPs and to determine if the two NRs of Fap1 play a role in bacterial aggregation. This is especially true given that the NR domain of SraP has a pI of 5.6, in contrast to the basic NRs of GspB (9.5 pI) and PsrP (9.9 pI) [47].
In summary, we have described for the first time the presence of a pneumococcal biofilm-like structure in the lungs of infected mice. We have determined that PsrP mediates a more intimate bacterium to bacterium interaction that contributes to the presence of large bacteria aggregates in vivo and increased biofilm biomass and aggregates in vitro. This property appears to be shared among other SRRPs including those of medically relevant bacteria such as S. aureus and S. gordonii, suggesting that it is a conserved function for this class of proteins. How these interactions contribute to pathogenesis remains to be fully determined, however, studies with other bacteria indicate that biofilms serve to inhibit phagocytosis, protect against defensin-mediated killing, and serve as a focal point of infection during early stages of disease. Future experiments will be required to determine the extent to which this may apply for SRRP-mediated aggregates in vivo.
Methods
Bacterial strains and media
Wild type strains used in this study included S. pneumoniae strain TIGR4 and the previously described clinical isolates IPD-5, TNE-6012, and TBE-6050 [8], [12], [14]. T4R is an unencapsulated derivative of TIGR4 [48]. S. aureus ISP479C and S. gordonii M99 and their corresponding isogenic mutants ISP479C ΔsraP, and M99 ΔgspB have also been previously described [17], [27]. All of the S. pneumoniae mutants used in this study including T4 ΔpsrP, T4 ΩpsrP-secY2A2, T4 ΩpsrP, and T4R ΩpsrP have been shown not to have polar effects on upstream and downstream gene transcription [12], [13]. S. pneumoniae and S. gordonii were grown in Todd-Hewitt broth (THB) or on blood agar plates at 37°C in 5% CO2. S. aureus were grown in Tryptic-Soy Broth (TSB) or on blood agar plates at 37°C. Stocks for the PsrP mutants were grown in media supplemented with 1 µg/mL of erythromycin, those complemented with the expression vector pNE1 were grown on media supplemented with 250 µg/mL of spectinomycin. SraP and GspB mutant stocks were grown in media supplemented with either 15 µg/mL of erythromycin or 5 µg/mL chloramphenicol respectively. E. coli strain DH5α (Invitrogen, Carlsbad CA) expressing recombinant PsrP constructs were grown with 50 µg/mL of kanamycin. Recombinant proteins were purified as previously described [13], [26]. To avoid stress effects on the bacteria, no antibiotics were added to the media during any of the experiments.
Infection of mice and collection of tissues
Female BALB/cJ mice, 5–6 weeks old, were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice were anesthetized with 2.5% vaporized isoflurane prior to challenge. Exponential phase cultures of S. pneumoniae were centrifuged, washed, and suspended in sterile phosphate buffered saline (PBS). For each experimental cohort at least 6 mice were instilled with either 107 cfu of TIGR4 or T4 ΔpsrP in 20 µL of PBS into the left nostril. After two days mice were sacrificed for tissue collection. For imaging experiments the intact lungs were collected and processed as described below. For enumeration of bacterial aggregates, nasal lavage fluid was collected from anesthetized mice by instillation and retraction of 20 µl PBS. The same mice were subsequently asphyxiated with compressed CO2, and BAL fluid collected by flushing the lungs twice with 0.5 ml of PBS using a sterile catheter. All animal experimentation was conducted following the National Institutes for Health guidelines for housing and care of laboratory animals. Animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee at The University of Texas Health Science Center at San Antonio.
Scanning electron microscopy (SEM)
Lungs were cut in a sagital orientation, fixed for 2 hours with 2.5% glutaraldehyde in PBS, and then rinsed twice for 3 min in 0.1 M phosphate buffer (pH 7.4). Lungs were submerged in 1% osmium diluted in Zetterquist's Buffer for 30 minutes then washed with the same buffer for 2 minutes [49]. This was followed by step-wise dehydration with ethanol (i.e. 70%, 95%, and 100%); the first two steps for 15 minutes, the last for 30 minutes. Samples were treated with hexamethyldisilizane for 5 minutes prior to drying in a desiccator overnight. The next day samples were sputter coated with gold palladium and viewed with a JEOL-6610 scanning electron microscope.
Visualization of bacterial aggregates ex vivo
From each mouse BAL and 1∶10 PBS diluted nasopharyngeal lavage elutes were smeared onto glass slides, heat fixed, and Gram-stained. Since the nasopharyngeal samples were mucoid, dilution of the samples was warranted. Bacteria were visualized using a CKX41 Olympus microscope at 200× magnification. For each biological sample 100 CFU were randomly selected, taking note of the approximate number of diplococci composing each CFU, either 1, 2–10, or >10. Images of the bacteria were acquired at 400× magnification to better show the multiple bacteria composing the aggregates.
Fluorescent microscopy of tissue sections
Lung tissues were excised and frozen in Tissue Tek O.C.T solution (Miles Scientific). 5 µm thick lung sections were cut at the University of Texas at San Antonio Histopathology Core and stored at −80° C. Bacteria in the lung sections were detected by immunofluorescence using antibody against the capsular polysaccharide. Sections were thawed, fixed with ice-cold acetone for 20 minutes, and then rehydrated with 70% ethyl alcohol and then PBS. Samples were permeabilized with 0.1% Triton-X-100 for 5 minutes then blocked with 10% fetal bovine serum (FBS) in F12 media for 1 hour. Sections were incubated with 1∶1,000 rabbit anti-serotype 4 pneumococcus antiserum (Statens Serum Institut, Denmark) overnight at 4°C. After washing for three times with 0.5% Tween-PBS, sections were covered with FBS-F12 containing goat anti-rabbit FITC conjugated antibody (Invitrogen) at 1∶2,000 and DAPI (5 µg/ml; for DNA) and the sections incubated for 1 hour at room temperature. Tissue sections were washed and mounted with FluorSave (Merck Biosciences). Images were acquired at 1,000× using a Nikon AX-70 fluorescent Microscope and images processed with SimplePCI software.
Visualization of early and mature biofilms
Early biofilm formation was examined by measuring the ability of cells to adhere and accumulate biomass on the bottom of a 96-well (flat-bottom) polystyrene plates (Costar, Corning Incorporated, Lowell MA) [24]. Microtiter wells with 200 µl THB were inoculated with 106 CFU of S. pneumoniae taken from cultures at mid-logarithmic phase growth (OD620 = 0.5). Plates were incubated at 37° C in 5% CO2. S. aureus and S. gordonii biofilm formation on microtiter plates was done in a similar manner, with the exception that TSB was used for S. aureus [50], [51]. Bacteria were grown for 2, 4, 6, 8, 18, and 24 h, after which the biofilms were washed gently with PBS and stained with 100 µL of 0.1% CV. Biofilm biomass was subsequently quantified by image capture using an inverted microscope at 15× and 100× magnification and measuring the corresponding optical density (A540) of the supernatant following washing of the bacteria and solubilization of CV in 200 µL of 95% ethanol.
Mature S. pneumoniae biofilms were grown under once through conditions in a glass slide chamber using a continuous-flow through reactor [25]. The flow cell was constructed of anodized aluminum containing a chamber (4.0 mm by 1.3 cm by 5.0 cm) having two glass surfaces, one being a microscope slide and the other being a glass coverslip serving as the substratum. S. pneumoniae cells grown to mid-logarithmic phase served as the inoculum and were injected into a septum 4 cm upstream from the flow cell. Bacteria were allowed to attach to the glass substratum for 2 hours prior to initiating flow. The flow rate of the system was adjusted to 0.014 ml/min. Flow through the chamber was laminar, with a Reynolds number of <0.5, having a fluid residence time of 180 min. Biofilms were grown at 37°C in 5% CO2 for 3 days under once through conditions. Biofilms were then visualized by confocal laser microscopy as described below.
Biofilms were also grown on the interior surface of a 1-meter long, size 16 Masterflex silicone tubing (0.89mm Internal Diameter, Cole Parmer Inc.) using once-through continuous flow conditions. The line was inoculated with 5 mL of a mid-logarithmic culture and the bacteria were allowed to attach for 2 hours. The flow rate of the system was adjusted to 0.035 ml/min and bacteria were grown for 3 days at 37°C in 5% CO2. Bacterial cells were harvested from the interior surface by pinching the tube along its entire length, resulting in removal of the cell material from the lumen of the tubing. Following extraction, exudates were gently suspended in 1 ml of PBS and the optical density (OD620) was measured. For light microscopy pictures, 50 µl of line exudate in saline was stained by the addition of 50 µL of 1% CV. A volume of 5 µl of stained line exudates was applied to glass slides, coverslipped, and images taken at 200× magnification using a light microscope. Viable cell counts were determined by plating serial dilutions of exudates following the disruption of each sample by vortexing. Biofilm biomass was determined by measuring the total protein concentration of the exudates by BCA following the complete lysis of S. pneumoniae with saline containing 0.1% deoxycholate and 0.1% sodium-dodecyl sulfate, which activates the murein hydrolase autolysin, or use of French press for S. gordonii and S. aureus cultures. For studies testing whether antibodies or recombinant protein inhibited bacteria aggregation media was supplemented with BR antiserum at 1∶1,000 or spiked with recombinant protein at a final concentration of 1.0 µM.
Confocal scanning laser microscopy and image acquisition
Confocal scanning laser microscopy was performed with an LSM 510 Meta inverted microscope (Zeiss, Heidelberg, Germany). Images were obtained with an LD-Apochrome 40×/0.6 lens and the LSM 510 Meta image acquisition software (Zeiss). To visualize the biofilm architecture of 3-day-old biofilms, biofilms were stained using the Live/Dead BacLight stain from Invitrogen (Carlsbad, CA). Quantitative analysis of epifluorescence microscopic images obtained from flow cell-grown biofilms at the 6-day time point was performed with COMSTAT image analysis software [52].
Far Western analysis of BR interactions
Recombinant full-length BR and truncated versions (BR.A, BR.B, BR.C) were expressed and purified from E. coli as previously described [13]. Glycosylated PsrPSRR2(33)-HIS was purified in the same manner from TIGR4 (Figure S3), with the exception that cultures were induced with 1% fucose and lysed with 1% SDS in PBS. Far Western analysis was carried out as described by Takamatsu et al. with minor modifications [53]. Nitrocellulose membranes were spotted with either 1 µg of whole cell lysate of S. pneumoniae, S. gordonii, S. aureus or E. coli expressing various PsrP constructs or with purified recombinant proteins in PBS. Membranes were incubated overnight in PBS with 4% bovine serum albumin and 0.1% Tween 20 (T-PBS) at room temperature. The next day, membranes were washed with T-PBS three times for 5 minutes, and incubated overnight at 4°C on an orbital platform rocker with T-PBS containing 1% bovine serum albumin (TB-PBS) with 1 µg/mL of Gst-BR, PsrPSRR2(33)-HIS, or the designated NR constructs from S. gordonii and S. aureus. Membranes were washed and incubated with monoclonal mouse anti-Gst antibody (1∶5,000 dilution) (Proto-Tech) overnight at 4°C in TB-PBS. Antibody binding was detected by incubating the membranes for 1 h with HRP-conjugated anti-mouse IgG (1∶10,000 dilution) (Sigma), followed by development with the Super Signal chemiluminescent detection system (Thermo Scientific). As a control for inadvertent interactions with the Gst tag, Far Western blots were also performed using an unrelated Gst-tagged Chlamydia trachomatis protein (TC0109; Figure S4). No interactions were observed.
Co-immunoprecipitation of Gst-BR with rBR
Co-immunoprecipitation of Gst-BR with the truncated versions of rPsrP was carried out as previously described by Shivshankar et al. [13]. Protein G Sepharose beads (Amersham) were incubated overnight at 4°C with mouse monoclonal penta-His antibody (1∶50; Qiagen) in 500 ml of F12 media supplemented with 10% fetal bovine serum. Beads were incubated with 400 µl of whole bacterial lysates from E. coli expressing penta-His tagged recombinant versions of PsrP spiked with 200 µg of recombinant Gst-BR full length and incubated overnight at 4°C with gentle agitation. Beads were washed with RIPA buffer, then boiled in sample buffer for 10 min [54]. Samples were separated on 12% SDS-PAGE gels and electrophoretically transferred to nitrocellulose membranes. Membranes were blocked with T-PBS containing 4% bovine serum for 30 min at room temperature. Membranes were then incubated overnight at 4°C with mouse anti-Gst (1∶7500; Proto-tech) in blocking buffer. Following incubation, membranes were washed with T-PBS three times for 5 minutes. HRP-conjugated goat anti-rabbit Immunoglobulin G (1∶10 000; Sigma) was used as the secondary antibody, followed by development with the Super Signal chemiluminescent detection system (Thermo Scientific).
Visualization of recombinant BR bound to TIGR4
For labeling of bacteria, TIGR4 and T4 ΔpsrP were pelleted and suspended in 1 ml of carbonate buffer (pH 9.0) containing FITC (1 mg/ml) and incubated in the dark at room temperature with constant end-to-end tumbling. FITC-labeled bacteria were washed with PBS (pH 7.4) and centrifuged, until the supernatant became clear. rBR fragments were labeled using a FluorLink-Ab Cy3 labeling kit (Amersham) using the instructions provided by the manufacturer. Labeled bacteria were suspended in serum-free F12 media containing the labeled constructs for 1 hour and gently mixed. Subsequently, pneumococci were washed and suspended in F12 medium. Labeled bacteria and bound recombinant protein were visualized using an AX-70 fluorescent microscope and the images were captured at 0.1112–0.8886 ms exposure time for Cy2 and Cy3 filters. The magnification used for capture of digital images was 1000×. Captured images were processed using Simple PCI software.
Adhesion assays
A549 cells (human alveolar type II pneumocytes; ATCC CRL-185), were grown to 90% confluence on 24-well plates (∼106 cells/well). Prior to use, cells were washed with cell F12 media to remove serum. For competitive inhibition binding assays, A549 cells were incubated with 1µM of either rBR, rBR.C, a synthesized peptide corresponding to AA 122–167, or BSA for 1 HR. Following incubation, cells were exposed to media that contained 107 cfu/mL of bacteria and incubated for 1 h at 37°C in 5% CO2. Nonadhering bacteria were removed by washing the cells 3 times with T-PBS and the number of adhering bacteria was determined by lysis of the monolayer with 0.1% Triton X-100 and plating wells per experiment.
Opsonophagocytosis assay
Bacterial cultures were centrifuged and suspended in 0.1M sodium carbonate buffer (pH 8.0) at an OD620 of 0.2. Care was taken to cause minimal disruption of the biofilm aggregates. The diluted cultures were labeled with fluorescent isothiocyanate (1mg/ml) for 30 min at room temperature in the dark. Following labeling, cultures were gently washed three times with sterile PBS to remove free FITC and suspended in PBS. FITC-labeled bacteria were opsonized with 3% control rabbit serum for 30 minutes at 37°C with mild periodical tapping. Mouse J774.1, macrophage cultures maintained in 10% FBS containing DMEM were used for phagocytosis of the opsonized pneumococci. Macrophages were harvested, washed and diluted with opsonophagocytosis buffer (PBS containing 0.2% BSA). FITC-labeled bacteria in 100 µl were added to 106 macrophage cells in 400 µl and incubated for 1 hour at 37°C with periodic shaking. Afterwards, the macrophages were pelleted and washed twice in the assay buffer. Cells were suspended in 400µl of 2% paraformaldehyde until flow cytometric analysis. A2-Laser BD FACSCaliber Analyzer (Becton Dickinson, NJ; Institutional Flow Cytometry Core Facility at the Health Science Center) was employed to analyze percent phagocytic uptake of the labeled bacteria by the macrophages. A minimum of 20,000 events were counted for each sample at 480 nm excitation and 530nm detection wavelengths. Background fluorescence was nullified by subjecting negative control macrophages in assay buffer without any fluorescent bacteria to FACS analysis. Data were processed using CellQuest software.
Statistical analysis
For pair-wise comparisons of groups statistical analyses were performed using a Student's t-test. For multivariate analyses a 1-Way ANOVA followed by a post-priori test using Sigma Stat software was used.
Supporting Information
Zdroje
1. 1999 Pneumococcal vaccines. WHO position paper. Wkly Epidemiol Rec 74 177 183
2. LexauCA
LynfieldR
DanilaR
PilishviliT
FacklamR
2005 Changing epidemiology of invasive pneumococcal disease among older adults in the era of pediatric pneumococcal conjugate vaccine. JAMA 294 2043 2051
3. OverturfG
PowarsD
1980 Infections in sickle cell anemia: pathogenesis and control. Tex Rep Biol Med 40 283 292
4. WongWY
OverturfGD
PowarsDR
1992 Infection caused by Streptococcus pneumoniae in children with sickle cell disease: epidemiology, immunologic mechanisms, prophylaxis, and vaccination. Clin Infect Dis 14 1124 1136
5. RoushSW
MurphyTV
2007 Historical comparisons of morbidity and mortality for vaccine-preventable diseases in the United States. JAMA 298 2155 2163
6. O'BrienKL
WolfsonLJ
WattJP
HenkleE
Deloria-KnollM
2009 Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet 374 893 902
7. MaruyamaT
GabazzaEC
MorserJ
TakagiT
D'Alessandro-GabazzaC
2010 Community-acquired pneumonia and nursing home-acquired pneumonia in the very elderly patients. Respir Med 104 584 592
8. ObertC
SublettJ
KaushalD
HinojosaE
BartonT
2006 Identification of a candidate Streptococcus pneumoniae core genome and regions of diversity correlated with invasive pneumococcal disease. Infect Immun 74 4766 4777
9. OrihuelaCJ
2009 Role played by psrP-secY2A2 (accessory region 34) in the invasive disease potential of Streptococcus pneumoniae. J Infect Dis 200 1180 1181; author reply 1181–1182
10. HavaDL
CamilliA
2002 Large-scale identification of serotype 4 Streptococcus pneumoniae virulence factors. Mol Microbiol 45 1389 1406
11. EmbryA
HinojosaE
OrihuelaCJ
2007 Regions of Diversity 8, 9 and 13 contribute to Streptococcus pneumoniae virulence. BMC Microbiol 7 80
12. RoseL
ShivshankarP
HinojosaE
RodriguezA
SanchezCJ
2008 Antibodies against PsrP, a novel Streptococcus pneumoniae adhesin, block adhesion and protect mice against pneumococcal challenge. J Infect Dis 198 375 383
13. ShivshankarP
SanchezC
RoseLF
OrihuelaCJ
2009 The Streptococcus pneumoniae adhesin PsrP binds to Keratin 10 on lung cells. Mol Microbiol 73 663 679
14. TettelinH
NelsonKE
PaulsenIT
EisenJA
ReadTD
2001 Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 293 498 506
15. TakamatsuD
BensingBA
SullamPM
2005 Two additional components of the accessory Sec system mediating export of the Streptococcus gordonii platelet-binding protein GspB. J Bacteriol 187 3878 3883
16. TakamatsuD
BensingBA
SullamPM
2004 Four proteins encoded in the gspB-secY2A2 operon of Streptococcus gordonii mediate the intracellular glycosylation of the platelet-binding protein GspB. J Bacteriol 186 7100 7111
17. BensingBA
GibsonBW
SullamPM
2004 The Streptococcus gordonii platelet binding protein GspB undergoes glycosylation independently of export. J Bacteriol 186 638 645
18. TakamatsuD
BensingBA
SullamPM
2004 Genes in the accessory Sec locus of Streptococcus gordonii have three functionally distinct effects on the expression of the platelet-binding protein GspB. Mol Microbiol 52 189 203
19. HammerschmidtS
WolffS
HockeA
RosseauS
MullerE
2005 Illustration of pneumococcal polysaccharide capsule during adherence and invasion of epithelial cells. Infect Immun 73 4653 4667
20. MoscosoM
GarciaE
LopezR
2009 Pneumococcal biofilms. Int Microbiol 12 77 85
21. Hall-StoodleyL
StoodleyP
2009 Evolving concepts in biofilm infections. Cell Microbiol 11 1034 1043
22. Hall-StoodleyL
HuFZ
GiesekeA
NisticoL
NguyenD
2006 Direct detection of bacterial biofilms on the middle-ear mucosa of children with chronic otitis media. JAMA 296 202 211
23. ReidSD
HongW
DewKE
WinnDR
PangB
2009 Streptococcus pneumoniae forms surface-attached communities in the middle ear of experimentally infected chinchillas. J Infect Dis 199 786 794
24. LizcanoA
ChinT
SauerK
TuomanenEI
OrihuelaCJ
2010 Early biofilm formation on microtiter plates is not correlated with the invasive disease potential of Streptococcus pneumoniae. Microb Pathog 48 124 130
25. AllegrucciM
HuFZ
ShenK
HayesJ
EhrlichGD
2006 Phenotypic characterization of Streptococcus pneumoniae biofilm development. J Bacteriol 188 2325 2335
26. TakamatsuD
BensingBA
ChengH
JarvisGA
SibooIR
2005 Binding of the Streptococcus gordonii surface glycoproteins GspB and Hsa to specific carbohydrate structures on platelet membrane glycoprotein Ibalpha. Mol Microbiol 58 380 392
27. SibooIR
ChambersHF
SullamPM
2005 Role of SraP, a Serine-Rich Surface Protein of Staphylococcus aureus, in binding to human platelets. Infect Immun 73 2273 2280
28. XiongYQ
BensingBA
BayerAS
ChambersHF
SullamPM
2008 Role of the serine-rich surface glycoprotein GspB of Streptococcus gordonii in the pathogenesis of infective endocarditis. Microb Pathog 45 297 301
29. SamenU
EikmannsBJ
ReinscheidDJ
BorgesF
2007 The surface protein Srr-1 of Streptococcus agalactiae binds human keratin 4 and promotes adherence to epithelial HEp-2 cells. Infect Immun 75 5405 5414
30. van SorgeNM
QuachD
GurneyMA
SullamPM
NizetV
2009 The Group B Streptococcal serine-rich repeat 1 glycoprotein mediates penetration of the blood-brain barrier. J Infect Dis 199 1479 1487
31. FroeligerEH
Fives-TaylorP
2001 Streptococcus parasanguis fimbria-associated adhesin Fap1 is required for biofilm formation. Infect Immun 69 2512 2519
32. HandleyPS
CorreiaFF
RussellK
RosanB
DiRienzoJM
2005 Association of a novel high molecular weight, serine-rich protein (SrpA) with fibril-mediated adhesion of the oral biofilm bacterium Streptococcus cristatus. Oral Microbiol Immunol 20 131 140
33. Konto-GhiorghiY
MaireyE
MalletA
DumenilG
CaliotE
2009 Dual role for pilus in adherence to epithelial cells and biofilm formation in Streptococcus agalactiae. PLoS Pathog 5 e1000422
34. ManettiAG
ZingarettiC
FalugiF
CapoS
BombaciM
2007 Streptococcus pyogenes pili promote pharyngeal cell adhesion and biofilm formation. Mol Microbiol 64 968 983
35. HelaineS
CarbonnelleE
ProuvensierL
BerettiJL
NassifX
2005 PilX, a pilus-associated protein essential for bacterial aggregation, is a key to pilus-facilitated attachment of Neisseria meningitidis to human cells. Mol Microbiol 55 65 77
36. Munoz-EliasEJ
MarcanoJ
CamilliA
2008 Isolation of Streptococcus pneumoniae biofilm mutants and their characterization during nasopharyngeal colonization. Infect Immun 76 5049 5061
37. ParkerD
SoongG
PlanetP
BrowerJ
RatnerAJ
2009 The NanA neuraminidase of Streptococcus pneumoniae is involved in biofilm formation. Infect Immun 77 3722 3730
38. TrappettiC
KadiogluA
CarterM
HayreJ
IannelliF
2009 Sialic acid: a preventable signal for pneumococcal biofilm formation, colonization, and invasion of the host. J Infect Dis 199 1497 1505
39. SoongG
MuirA
GomezMI
WaksJ
ReddyB
2006 Bacterial neuraminidase facilitates mucosal infection by participating in biofilm production. J Clin Invest 116 2297 2305
40. OggioniMR
TrappettiC
KadiogluA
CassoneM
IannelliF
2006 Switch from planktonic to sessile life: a major event in pneumococcal pathogenesis. Mol Microbiol 61 1196 1210
41. AspirasMB
EllenRP
CvitkovitchDG
2004 ComX activity of Streptococcus mutans growing in biofilms. FEMS Microbiol Lett 238 167 174
42. AllegrucciM
SauerK
2007 Characterization of colony morphology variants isolated from Streptococcus pneumoniae biofilms. J Bacteriol 189 2030 2038
43. MoscosoM
GarciaE
LopezR
2006 Biofilm formation by Streptococcus pneumoniae: Role of choline, extracellular DNA, and capsular polysaccharide in microbial accretion. J Bacteriol 188 7785 7795
44. OrihuelaCJ
RadinJN
SublettJE
GaoG
KaushalD
2004 Microarray analysis of pneumococcal gene expression during invasive disease. Infect Immun 72 5582 5596
45. WuH
ZengM
Fives-TaylorP
2007 The glycan moieties and the N-terminal polypeptide backbone of a fimbria-associated adhesin, Fap1, play distinct roles in the biofilm development of Streptococcus parasanguinis. Infect Immun 75 2181 2188
46. StephensonAE
WuH
NovakJ
TomanaM
MintzK
2002 The Fap1 fimbrial adhesin is a glycoprotein: Antibodies specific for the glycan moiety block the adhesion of Streptococcus parasanguis in an in vitro tooth model. Mol Microbiol 43 147 157
47. ZhouM
WuH
2009 Glycosylation and biogenesis of a family of serine-rich bacterial adhesins. Microbiology 155 317 327
48. MannB
OrihuelaC
AntikainenJ
GaoG
SublettJ
2006 Multifunctional role of Choline binding protein G in pneumococcal pathogenesis. Infect Immun 74 821 829
49. SheldonH
ZetterquistH
1955 Internal ultrastructure in granules of white blood cells of the mouse; a preliminary note. Bull Johns Hopkins Hosp 96 135 139
50. IzanoEA
AmaranteMA
KherWB
KaplanJB
2008 Differential roles of poly-N-acetylglucosamine surface polysaccharide and extracellular DNA in Staphylococcus aureus and Staphylococcus epidermidis biofilms. Appl Environ Microbiol 74 470 476
51. LooCY
CorlissDA
GaneshkumarN
2000 Streptococcus gordonii biofilm formation: identification of genes that code for biofilm phenotypes. J Bacteriol 182 1374 1382
52. HeydornA
NielsenAT
HentzerM
SternbergC
GivskovM
2000 Quantification of biofilm structures by the novel computer program COMSTAT. Microbiology 146 (Pt 10) 2395 2407
53. TakamatsuD
BensingBA
PrakobpholA
FisherSJ
SullamPM
2006 Binding of the Streptococcal surface glycoproteins GspB and Hsa to human salivary proteins. Infect Immun 74 1933 1940
54. AusubelFM
BrentR
KingstonRE
MooreDD
SeidmanJG
2008 Current Protocols in Molecular Biology Hoboken, NJ John Wiley & Sons, Inc
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 8
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- The Transcription Factor Rbf1 Is the Master Regulator for -Mating Type Controlled Pathogenic Development in
- PKC Signaling Regulates Drug Resistance of the Fungal Pathogen via Circuitry Comprised of Mkc1, Calcineurin, and Hsp90
- Contribution of Coagulases towards Disease and Protective Immunity
- Early Severe Inflammatory Responses to Uropathogenic Predispose to Chronic and Recurrent Urinary Tract Infection