
Genome-Wide Gene-Environment Study Identifies Glutamate Receptor Gene as a Parkinson's Disease Modifier Gene via Interaction with Coffee
Our aim was to identify genes that influence the inverse association of coffee with the risk of developing Parkinson's disease (PD). We used genome-wide genotype data and lifetime caffeinated-coffee-consumption data on 1,458 persons with PD and 931 without PD from the NeuroGenetics Research Consortium (NGRC), and we performed a genome-wide association and interaction study (GWAIS), testing each SNP's main-effect plus its interaction with coffee, adjusting for sex, age, and two principal components. We then stratified subjects as heavy or light coffee-drinkers and performed genome-wide association study (GWAS) in each group. We replicated the most significant SNP. Finally, we imputed the NGRC dataset, increasing genomic coverage to examine the region of interest in detail. The primary analyses (GWAIS, GWAS, Replication) were performed using genotyped data. In GWAIS, the most significant signal came from rs4998386 and the neighboring SNPs in GRIN2A. GRIN2A encodes an NMDA-glutamate-receptor subunit and regulates excitatory neurotransmission in the brain. Achieving P2df = 10−6, GRIN2A surpassed all known PD susceptibility genes in significance in the GWAIS. In stratified GWAS, the GRIN2A signal was present in heavy coffee-drinkers (OR = 0.43; P = 6×10−7) but not in light coffee-drinkers. The a priori Replication hypothesis that “Among heavy coffee-drinkers, rs4998386_T carriers have lower PD risk than rs4998386_CC carriers” was confirmed: ORReplication = 0.59, PReplication = 10−3; ORPooled = 0.51, PPooled = 7×10−8. Compared to light coffee-drinkers with rs4998386_CC genotype, heavy coffee-drinkers with rs4998386_CC genotype had 18% lower risk (P = 3×10−3), whereas heavy coffee-drinkers with rs4998386_TC genotype had 59% lower risk (P = 6×10−13). Imputation revealed a block of SNPs that achieved P2df<5×10−8 in GWAIS, and OR = 0.41, P = 3×10−8 in heavy coffee-drinkers. This study is proof of concept that inclusion of environmental factors can help identify genes that are missed in GWAS. Both adenosine antagonists (caffeine-like) and glutamate antagonists (GRIN2A-related) are being tested in clinical trials for treatment of PD. GRIN2A may be a useful pharmacogenetic marker for subdividing individuals in clinical trials to determine which medications might work best for which patients.
Published in the journal:
. PLoS Genet 7(8): e32767. doi:10.1371/journal.pgen.1002237
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002237
Summary
Our aim was to identify genes that influence the inverse association of coffee with the risk of developing Parkinson's disease (PD). We used genome-wide genotype data and lifetime caffeinated-coffee-consumption data on 1,458 persons with PD and 931 without PD from the NeuroGenetics Research Consortium (NGRC), and we performed a genome-wide association and interaction study (GWAIS), testing each SNP's main-effect plus its interaction with coffee, adjusting for sex, age, and two principal components. We then stratified subjects as heavy or light coffee-drinkers and performed genome-wide association study (GWAS) in each group. We replicated the most significant SNP. Finally, we imputed the NGRC dataset, increasing genomic coverage to examine the region of interest in detail. The primary analyses (GWAIS, GWAS, Replication) were performed using genotyped data. In GWAIS, the most significant signal came from rs4998386 and the neighboring SNPs in GRIN2A. GRIN2A encodes an NMDA-glutamate-receptor subunit and regulates excitatory neurotransmission in the brain. Achieving P2df = 10−6, GRIN2A surpassed all known PD susceptibility genes in significance in the GWAIS. In stratified GWAS, the GRIN2A signal was present in heavy coffee-drinkers (OR = 0.43; P = 6×10−7) but not in light coffee-drinkers. The a priori Replication hypothesis that “Among heavy coffee-drinkers, rs4998386_T carriers have lower PD risk than rs4998386_CC carriers” was confirmed: ORReplication = 0.59, PReplication = 10−3; ORPooled = 0.51, PPooled = 7×10−8. Compared to light coffee-drinkers with rs4998386_CC genotype, heavy coffee-drinkers with rs4998386_CC genotype had 18% lower risk (P = 3×10−3), whereas heavy coffee-drinkers with rs4998386_TC genotype had 59% lower risk (P = 6×10−13). Imputation revealed a block of SNPs that achieved P2df<5×10−8 in GWAIS, and OR = 0.41, P = 3×10−8 in heavy coffee-drinkers. This study is proof of concept that inclusion of environmental factors can help identify genes that are missed in GWAS. Both adenosine antagonists (caffeine-like) and glutamate antagonists (GRIN2A-related) are being tested in clinical trials for treatment of PD. GRIN2A may be a useful pharmacogenetic marker for subdividing individuals in clinical trials to determine which medications might work best for which patients.
Introduction
Common disorders are thought to have both genetic and environmental components. Genome-wide association studies (GWAS) have successfully identified numerous susceptibility loci for many common disorders ranging from behavioral traits such as addiction and substance abuse to infectious and immune-related disorders, age-related neurodegenerative disorders like Alzheimer's, Parkinson's and macular degeneration, metabolic disorders, psychiatric disorders, and many more (for the list and results of over 800 published GWAS see http://www.genome.gov/gwastudies). Despite the success of GWAS, the heritability of common disorders cannot be fully explained by the genes that have been discovered [1]. GWAS are built on the notion that common alleles predispose to common disorders. Rare variants, which are probably responsible for some of the missing heritability, would not have been detected by GWAS. Sequencing the genome and novel analytical methods will help identify the rare variants. Another hiding place for the missing heritability is in interactions. Genes that impact disease through interactions with other genes or environmental factors are not detected by GWAS if their main effects are small. GWAS can only identify genes that exhibit significant main effects; genes that require the interacting factor to be included in the study to show their association with disease are missed. Inclusion of key environmental factors in genome-wide studies is anticipated to be an important next step for deciphering the genetic structure of common multifactorial disorders. Amassing sufficient analytic power for gene-environment studies, however, is a challenge. Power decreases dramatically as a function of frequency of exposure, number of parameters being estimated and sample size. Interaction studies require at least four times the sample size that standard GWAS would require to detect an effect of similar magnitude (reviewed in [2]). Yet, there are fewer datasets with both DNA and environmental exposure data than those with DNA alone, and their sample sizes are often smaller.
Parkinson's disease (PD) is a classic example of a common multifactorial disorder. PD is characterized by neurodegeneration in the substantia nigra that manifests initially as a movement disorder but often leads to cognitive and psychiatric problems as well. PD is progressive and there is no treatment currently available that could prevent or slow disease progression. PD is the second most common neurodegenerative disease after Alzheimer's disease; it affects about 5 million individuals in the 10 most populous nations and is expected to double in frequency by 2030 [3]. Until the 1990's PD was thought to be purely environmental with no genetic component. In the last decade, numerous genes have been identified, some of which can cause PD [4] and others that are susceptibility loci [5]–[10]. There are also compelling data from epidemiology that cigarette smoking and caffeinated-coffee consumption are associated with reduced risk of developing PD [11], [12] and that exposure to environmental neurotoxins is associated with increased risk of developing PD [13]. Thus PD is a strong candidate for studying gene-environment interactions [14].
We conducted a genome-wide association and interaction study (GWAIS) using the joint test [15] for each SNP's marginal association and its interaction with coffee consumption on PD risk, followed by stratified GWAS in heavy and light coffee drinkers (see Analytic Strategy in Materials and Methods section). Our aim was to identify genes that enhance or diminish the protective effect of caffeinated-coffee for use as biomarkers for pharmacogenetic prevention and treatment. Caffeine is an adenosine-receptor antagonist. In animal models of PD, where administration of neurotoxins is used to destroy dopaminergic neurons mimicking PD, caffeine and selective A2A-antagonists have been shown to be neuroprotective and attenuate dopamine loss [16]. Selective A2A-antagonists have been studied in human clinical trials and found to be safe, well tolerated and to provide symptomatic benefit for persons with PD [17], [18]; however, efficacy has not been high enough in the first generation of the drugs to meet regulatory approval for use as PD drugs. We posit that subsets of patients with certain genotypes may respond well to a given treatment and others may not. When they are combined the average efficacy may be insufficient for regulatory approval, while a subgroup of patients with certain genotype might still benefit substantially. If our prediction is correct, incorporating genetics in clinical trials of PD could revolutionize PD drug development. By examining the interaction of caffeinated-coffee with 811,597 SNPs in a hypothesis-free genome-wide study, we discovered GRIN2A as a novel PD modifier gene. GRIN2A encodes a subunit of the NMDA-glutamate-receptor which is well known for regulating excitatory neurotransmission in the brain and for controlling movement and behavior.
Materials and Methods
Human subjects
Human Subject Committees of the participating institutions approved the study. The Discovery dataset was nested in the NeuroGenetics Research Consortium (NGRC) GWAS which successfully identified known PD genes as well as a novel association with HLA [5] which has been widely replicated [10], [19]. For the present GWAIS, Replication samples were provided by PEG [20] (Parkinson, Environment, and Gene), PAGE [21] (Parkinson's, Genes, and Environment from the prospective NIH-AARP Diet and Health Study cohort), and HIHG [9] (Hussman Institute for Human Genomics). Persons with PD had been diagnosed by neurologists using standard criteria [22], control subjects self-reported as not having PD. Cases and controls were all unrelated, non-Hispanic Caucasian, from United States. The NGRC cohort was clinic-based sequentially ascertained patients, PEG and PAGE were community-based incident cases, HIHG was clinic-based and self-referral cases. The numbers of cases/controls with genotype, coffee/caffeine and key clinical and demographic data were NGRC = 1458/931, PEG = 280/310, PAGE = 525/1474, HIHG = 209/133 (Table S1).
Coffee/caffeine
NGRC, PEG and HIHG had collected lifetime caffeinated-coffee consumption data, measured as cups per day multiplied by the number of years of consumption (ccy) [12], [23]. PAGE had daily mg caffeine intake from all caffeine-containing drinks and foods for 12 months prior to enrollment (1995–1996) and only incident PD cases diagnosed after 1997 were included in the analysis [24]. Despite the variation in data collection, results were consistent across studies, corroborating robustness of the interaction between coffee/caffeine and GRIN2A. We could not, and did not, attempt to distinguish the bioactive ingredient in caffeinated-coffee. Although caffeine has been shown to be neuroprotective, there may be other ingredients in caffeinated-coffee that may affect disease pathogenesis. To classify coffee/caffeine intake, each dataset was treated separately according to the measurements available. The median ccy or mg was determined for controls within each dataset (excluding those with zero intake) and used as the cut-off for heavy drinkers (>median) vs. light drinkers (0 to ≤median). The median was 67.5 ccy for NGRC, 74.0 ccy for PEG, 70.0 ccy for HIHG, and 237.8 mg/day for PAGE. For coffee dose, quartiles were defined for each dataset using the full range from zero to maximum intake in controls. Results shown for NGRC, PEG and HIHG are based on lifetime caffeinated-coffee consumption. Truncating coffee use at age-at-onset or age-at-diagnosis in patients did not affect the results. To assess the effects of caffeinated tea and soda, we performed sensitivity analysis in NGRC dataset. Caffeinated soda and tea were commonly and equally consumed by heavy and light coffee drinkers (soda: 80% in both heavy and light drinkers; caffeinated tea: 66% in heavy coffee drinkers and 61% in light coffee drinkers). We repeated GWAIS and stratified GWAS with caffeinated soda and tea as covariates. We also explored association of caffeinated tea and soda with PD expecting an inverse association if caffeine were the bioactive ingredient in coffee.
Genotyping
The source of DNA was whole blood for NGRC and HIHG, saliva for PAGE, and whole blood (all PD and half of controls) or saliva (half of controls) for PEG. NGRC was genome-wide genotyped using Illumina HumanOmni1-Quad_v1-0_B array and achieved 99.92% call rate and 99.99% reproducibility. GWAS genotyping and statistical quality control (QC) have been published [5]. 811,597 SNPs (excluding Y chromosome SNPs because they are not amenable to sex adjustment) passed GWAS QC and were included in GWAIS. Replication groups genotyped GRIN2A_rs4998386. Only one SNP was genotyped for replication; we have no other undisclosed replication results. PEG and HIHG used ABI TaqMan assay-by-design (C__28018721_20), PAGE used Sequenom and all achieved call rates of 96%–99%.
Analytic strategy
The first step was to test the hypothesis that the effect of coffee on PD risk is affected by a gene; ie, test statistical interaction between SNPs and coffee genome-wide. Theoretically, a test of SNP*coffee interaction would have been suitable; however, a pure test of interaction has low power; reportedly, it requires more than four times the sample size that GWAS would require to detect a main effect of similar size (reviewed in [2]). We chose the joint test of SNP main effect and its interaction with coffee as proposed by Kraft et al [15]. We call the test GWAIS for genome-wide association and interaction study. The main advantage of the joint test is that it does test for interaction and it has more power than pure interaction test when there is a modest SNP marginal effect. Next we performed stratified GWAS in heavy and light drinkers to gain insight to where the interaction signal was coming from and to formulate a hypothesis for replication. We then replicated the top signal and performed pooled analysis. Methods for meta-analysis of the joint test are available [25], [26]; however, since we had individual level data we pooled the datasets.
Statistical analysis
Quality control for GWAIS and stratified GWAS in Discovery (NGRC)
The genome-wide genotypes for NGRC had been cleaned previously for GWAS using standard rigorous measures [5]. We had identified two significant principal components (PC1, PC2) marking Jewish/non-Jewish ancestry and European countries of origin [5]. Sex was a significant variable, because PD affects more men than women and our data has a significant gender disparity (Table S1). Controls were older than patients at age at onset, which was by design to minimize the chances that controls were too young to have developed the disease. Nevertheless, we controlled for age at enrolment both for patients and controls to avoid confounding by age-related factors. We examined coffee consumption and the most significant SNP for potential variation by disease related variables, recruitment sites, and ethnic and geographic origins of subjects (Table S2). Smoking was a potential confounder because it is correlated with coffee use and is an independent inverse risk factor of PD. Thus we repeated all analyses with smoking included as a covariate in the model (Table S3 without smoking as covariate, Table S4 with smoking as covariate). We also repeated analyses with caffeinated tea and soda in the model (Table S5). For details on how the data on tea, soda and smoking were collected in NGRC, see [12].
GWAIS in Discovery
811,597 SNP genotypes [5] and lifetime caffeinated-coffee consumption data [12] from 1458 persons with PD and 931 controls from NGRC were analyzed. We tested the following models: [SNP+coffee+SNP*coffee+covariate vs. coffee+covariate] henceforth referred to as [SNP+SNP*coffee] joint test [15]. Critically, the main effect of coffee on PD risk was present in both models being compared thus we controlled for coffee in the test. This model conducts a 2 degrees of freedom (df) joint test of SNP marginal effect and its interaction with coffee on PD risk [15]. Sex, age, PC1 and PC2 were included as covariates. We used likelihood ratio test statistics as implemented in PLINK [27], and tested the Dominant, Additive and Recessive modes of inheritance. GWAIS was repeated once with the addition of smoking as a covariate, and again by addition of caffeinated tea and soda as covariates.
Stratified GWAS in Discovery
There were 512 cases and 387 controls in the heavy coffee drinking group and 946 cases and 544 controls in the light coffee drinking group. We tested association of 811,597 SNPs with PD in each group using standard GWAS with 1 df. We used PLINK [27] and adjusted for age, sex, PC1 and PC2. Stratified GWAS were repeated with smoking, caffeinated soda and caffeinated tea added as covariates (Tables S4 and S5).
Replication
Based on the main finding in Discovery, we specified the replication hypothesis a-priori as follows: “Among heavy coffee drinkers, carriers of rs4998386_T allele have a lower risk of PD than carriers of rs4998386_CC genotype”. Note that we were using the GWAIS as a means to identify the genes that might enhance the inverse association of coffee with PD with the goal of carrying the discovery forward to pharmacogenetic studies. Hence, the replication hypothesis was framed as specified. We used three datasets for replication PEG [20], PAGE [21], and HIHG [9]. We tested between-study heterogeneity using Breslow-Day test statistics. There was no heterogeneity in coffee use, in rs4998386_CC or in rs4998386_TC genotype frequencies, but rs4998386_TT frequency, which is quite rare, varied significantly across studies. There were a total of 26 cases and 26 controls with rs4998386_TT genotype in Discovery and Replication combined. We found no trend in rs4998386_TT subject characteristics that could help pinpoint the source of heterogeneity (Table S6). Given the unanticipated heterogeneity in rs4998386_TT, we performed genotype-specific analysis (comparing TC to CC, excluding TT) as well as Dominant and Additive models which included TT. Categorical data were analyzed using logistic regression in SAS (version 9.2) and were adjusted for age and sex, and for source of data when data were pooled. Age at onset was analyzed as a continuous variable using linear regression in SAS.
Significance
P values were two-sided for Discovery, one-sided for Replication given the clear directional prior hypothesis [28], and two sided when Discovery and Replication were pooled. There is no agreed-upon significance threshold for GWAIS. The Bonferroni corrected threshold for all 811,597 SNPs on the array is P<6.4×10−8. However, not all 811,597 SNPs are independent due to linkage disequilibrium (LD). SimpleM [29] provides a sound Bonferroni-based multiple testing correction method for GWAS based on the estimated number of independent tests, allowing for marker-to-marker LD. It was shown to be the best approximation for permutation, which is computationally prohibitive for GWAS. Using simpleM we calculated the number of independent SNPs genome-wide for NGRC as Meff = 430,151; thus the Bonferroni corrected threshold for independent tests was P<1.16×10−7.
Imputation
We used IMPUTE v2 [30] with HapMap and 1000 Genomes genotypes combined as reference data to infer genotypes for SNPs that were not originally included on the Illumina OMNI-1 array and thus not genotyped in the NGRC dataset. 2,710,971 SNPs were imputed with high reliability (information score ≥0.95) and had MAF>0.01, increasing the genomic coverage to 3,522,568 SNPs total (genotyped and imputed). We performed GWAIS and stratified GWAS for the GRIN2A region (Chromosome 16, 97 Mb–102 Mb) using genotype probability data (dose 2-0) in R software http://www.r-project.org/.
Linkage disequilibrium
Linkage disequilibrium and haplotype blocks were estimated using the Haploview software [31]. Haplotype analysis was performed using hapstat adjusting for sex and age [32].
Copy number variations (CNV)
We used Golden Helix SNP Variation Suite version 7.2.3 (http://www.goldenhelix.com/) and PennCNV [33] to explore for deletions or duplications in the GRIN2A region. Golden Helix found no CNVs; PennCNV identified two controls with CNVs, which even if confirmed to be real, would not affect the results of the study.
Data access
NGRC genome-wide genotypes, phenotype and environmental data are available at www.ncbi.nlm.nih.gov/gap study accession number phs000196.v1.p1.
Results
GWAIS in Discovery
The most significant result was the novel appearance, on the Manhattan plot (Figure 1A, Figure S1), of a block of linked SNPs which map to the GRIN2A gene on chromosome 16 (Figure S2). This locus had not been detected in PD GWAS previously because its main effect is modest. However, when considered in the context of interaction with coffee, GRIN2A surpassed all known PD-associated genes in significance including SNCA which has been the strongest association with PD in GWAS. The signal for known PD genes were driven only by their main effects with no evidence for interaction (Pinteraction = 0.5–0.7); whereas the signals for PD-associated SNPs in GRIN2A were enhanced by SNP*coffee interaction (Pinteraction∼10−3). The quantile-quantile (QQ) plot of the expected vs. observed genome-wide P values (Figure 1B) is also evidence for the impact of GRIN2A on PD risk.
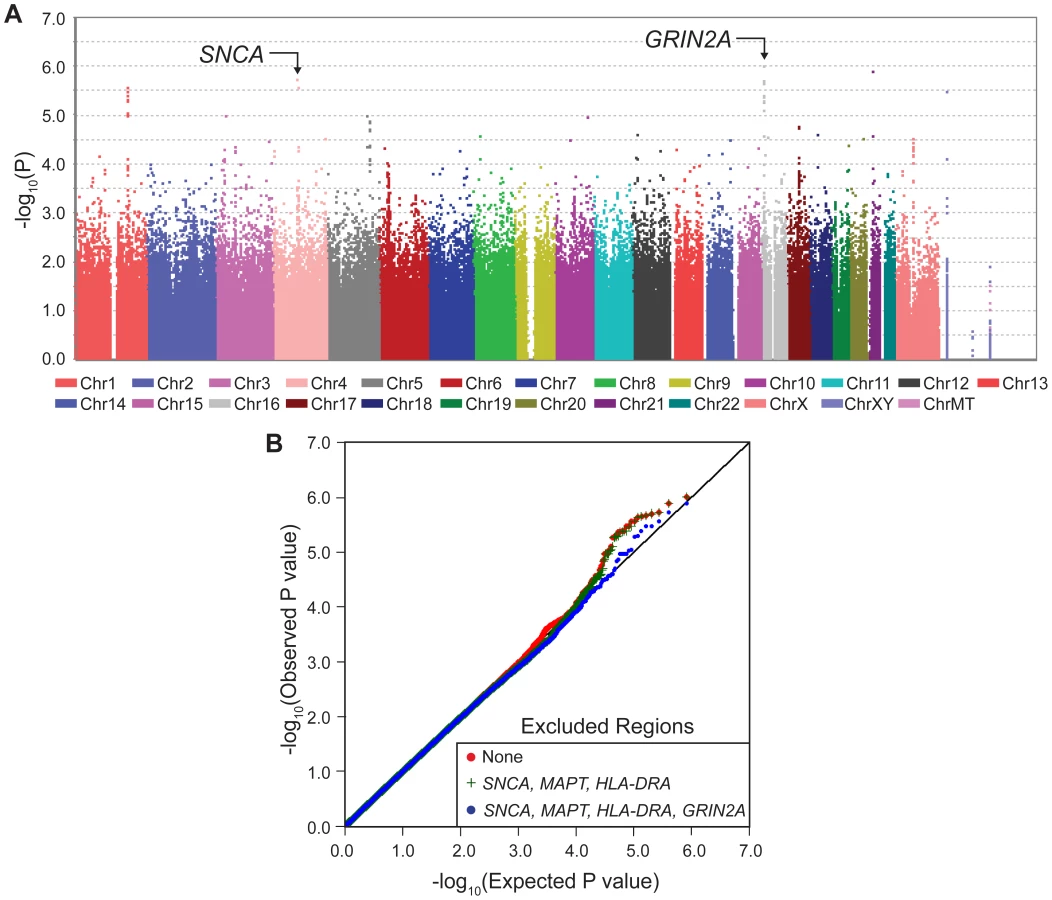
GWAIS results described above were obtained from a test that measures the combined significance of the SNP and its interaction with coffee on risk of PD [15]. The test has 2 df; hence when interaction is absent, GWAIS is less powerful than GWAS which has only 1 df. Furthermore, the sample size was smaller in GWAIS because it required not only genotypes but also coffee data, which was available for 2/3 of NGRC. Under these conditions, GWAIS produced P2df>10−6 (Figure 1A) for the top SNP in SNCA which had reached P = 3×10−11 in NGRC GWAS [5]. This drop in significance demonstrates the dramatic loss of power in GWAIS as compared to GWAS. Under these conditions, GWAIS yielded P2df = 1×10−6 for rs4998386 in GRIN2A (as compared to P2df = 3×10−6 for SNCA and P2df = 7×10−5 for MAPT). Dominant and Additive models produced nearly identical results for GRIN2A SNPs (Table 1). Recessive model had no notable signal (Figure S1).
GWAS in heavy and light coffee-drinkers in Discovery samples
With one goal being pharmacogenetic applications, we were interested in genes that modulate risk in people who consume caffeine, thus we stratified the subjects as heavy drinkers or light drinkers (light includes non-drinkers) and performed GWAS in each group (SNP-PD test, 1 df). The sample size was now further reduced to only 512 cases and 387 controls who drank more than the median (heavy drinkers) and 946 cases and 544 control subjects who drank less than the median (light drinkers). As expected due to interaction, which suggests different association patterns across categories, most of the signals seen in GWAIS (Figure 1A) appeared within either heavy drinkers (Figure 2, Table 2, Figure S1) or light drinkers (Figure 3, Table 2, Figure S1). In heavy drinkers, the focus of this study, the most significant result was GRIN2A_rs4998386 (P = 6×10−7) and 11 neighboring SNPs (P = 10−5 to 10−6, Table 2).
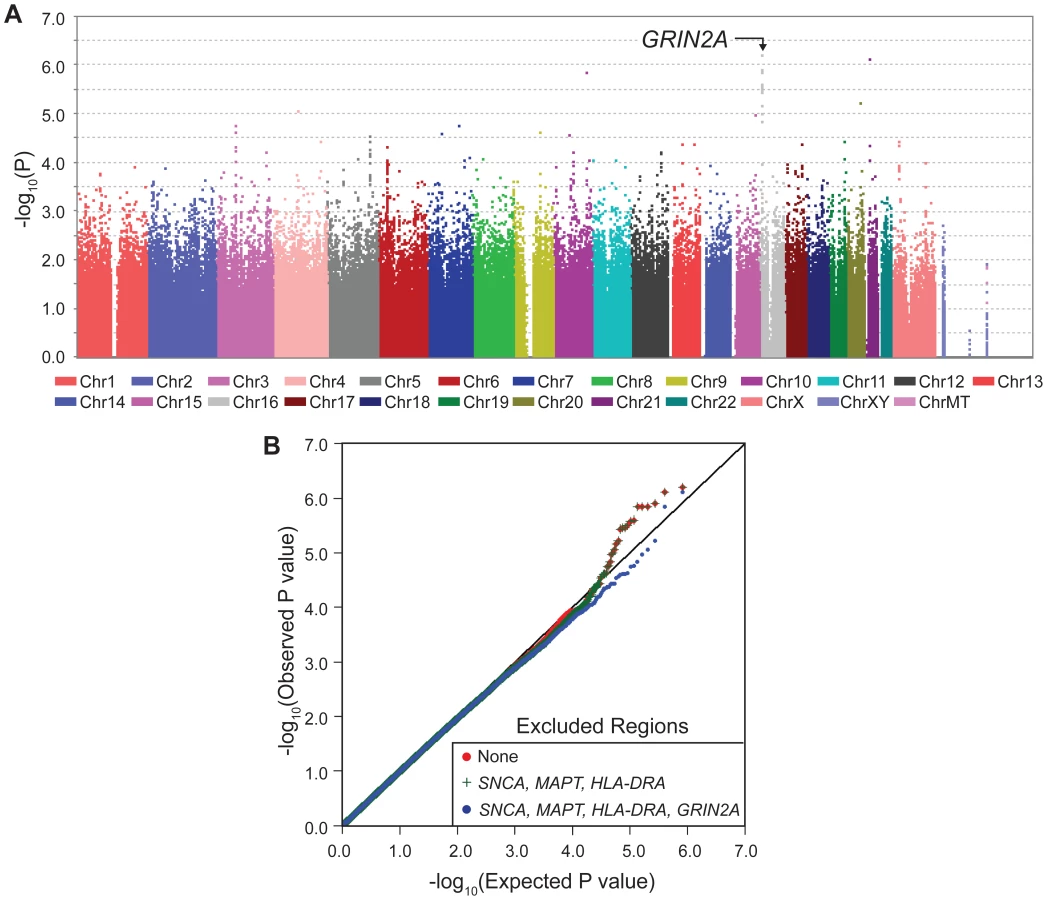
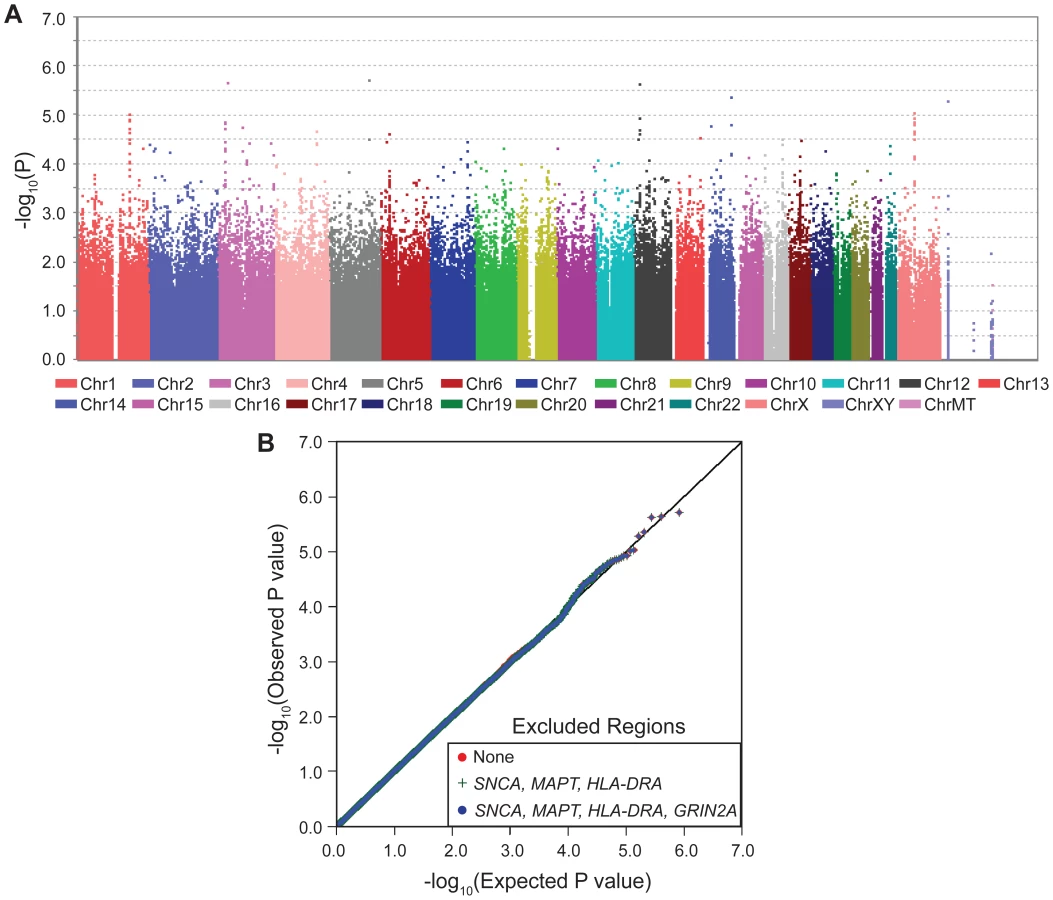
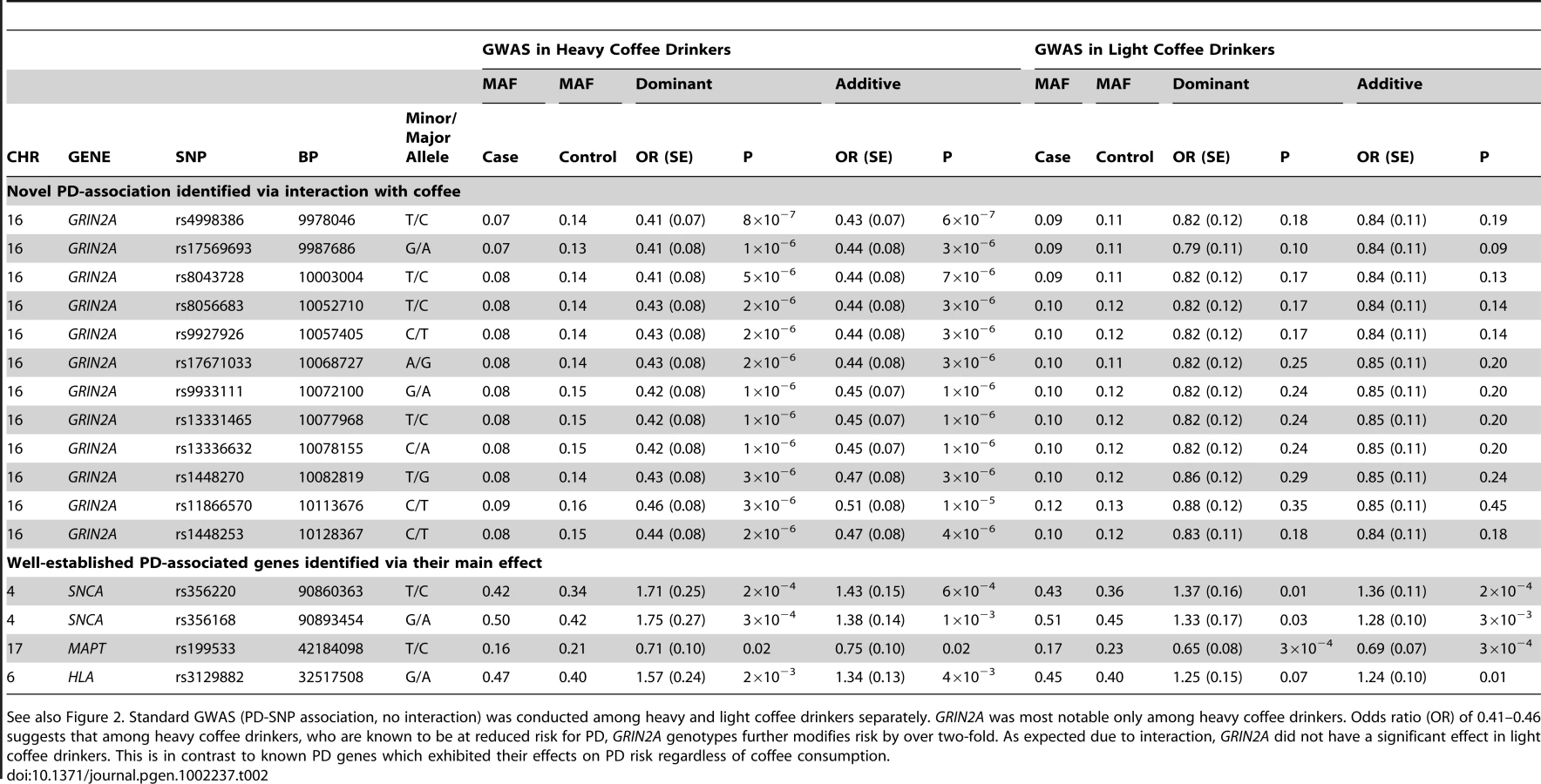
The QQ plots for stratified GWAS also demonstrate clearly that GRIN2A is the single primary PD associated locus in heavy coffee drinkers (Figure 2): exclusion of SNCA, HLA and MAPT did not have an impact in heavy drinkers, whereas exclusion of GRIN2A nearly abolished the extreme P values of 10−5–10−6. No clear signals were detected in light coffee drinkers (Figure 3).
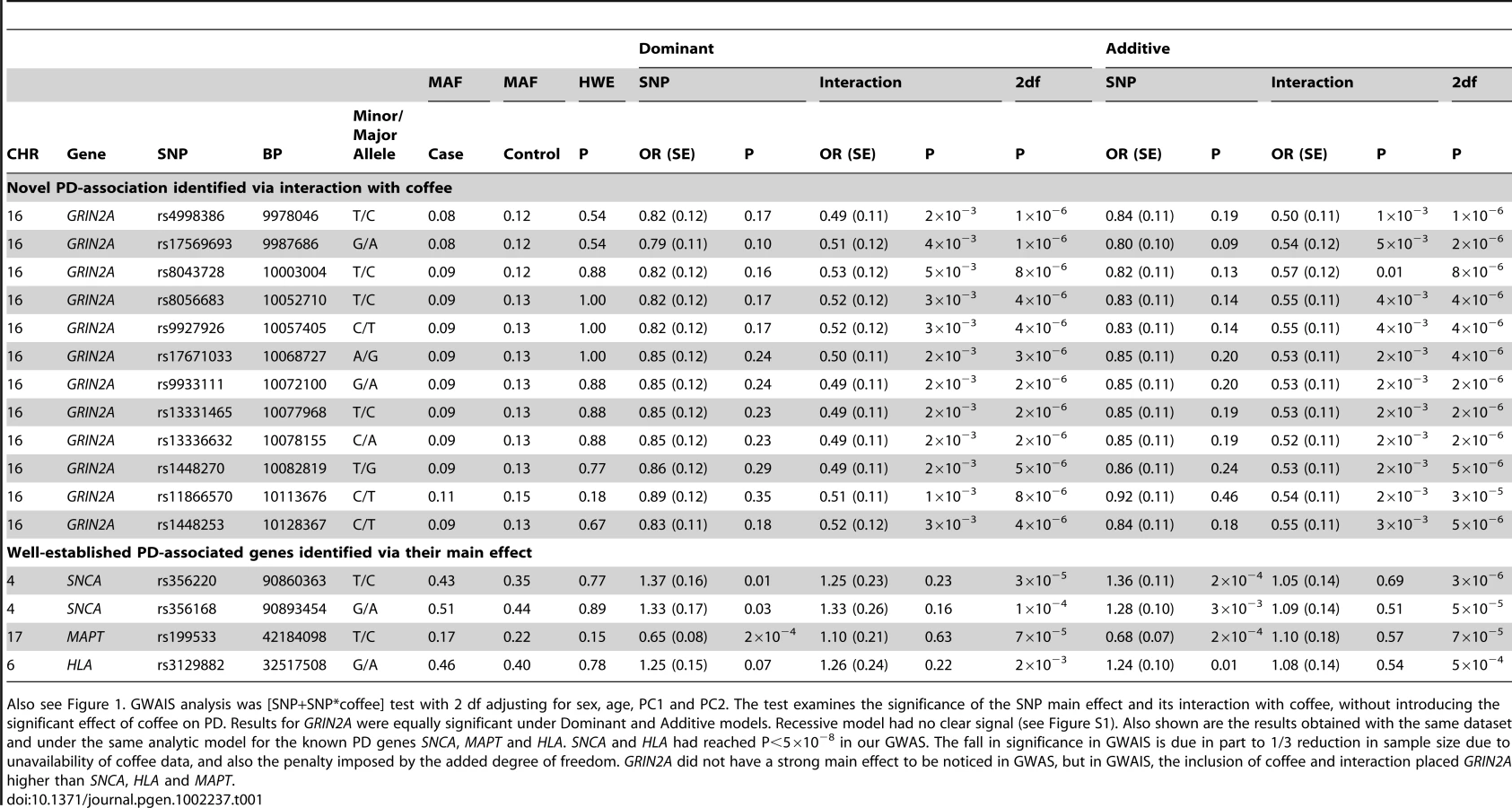
The 12 GRIN2A SNPs that were associated with PD via heavy coffee consumption had similar minor allele frequencies (MAF = 0.13–0.16 in controls) and odds ratios (OR = 0.43–0.51) and were in strong LD (Figure S3). Haplotype analysis did not strengthen the signal. Within this gene varying CNV software tools called either no CNVs or just two CNVs in controls. Thus, CNVs are unlikely to explain a large fraction of the phenotype variability. We therefore selected only the SNP with the lowest P value for replication (GRIN2A_rs4998386).
Genotype-specific association of coffee with PD in Discovery
Testing the association of coffee with PD in NGRC, when calculated irrespective of genotype, showed an average of 34% lower PD risk in heavy coffee drinkers than in light drinkers (OR = 0.66, P = 6×10−6, Table 3, Coffee irrespective of genotype). GRIN2A, irrespective of coffee, had a modest main effect on PD in NGRC (Table 3, GRIN2A rs4998386 genotype irrespective of coffee). A key question was if, and to what degree, GRIN2A_rs4998386 genotype modifies the effect of coffee on PD risk (Table 3): Within heavy drinkers, PD risk was 58% lower (OR = 0.42, P = 2×10−6) for rs4998386_TC, and 81% lower (OR = 0.19, P = 0.05) for rs4998386_TT genotype than rs4998386_CC; whereas in light drinkers genotype had no effect on risk. Similar results were obtained for Additive and Dominant models (Table S3). The joint effect comparing rs4998386_TC genotype and heavy coffee vs. rs4998386_CC genotype and light coffee was most dramatic, suggesting a highly significant 68% risk reduction (OR = 0.32, P = 7×10−11) in NGRC (Table 3, Joint effects of GRIN2A rs4998386 and coffee).
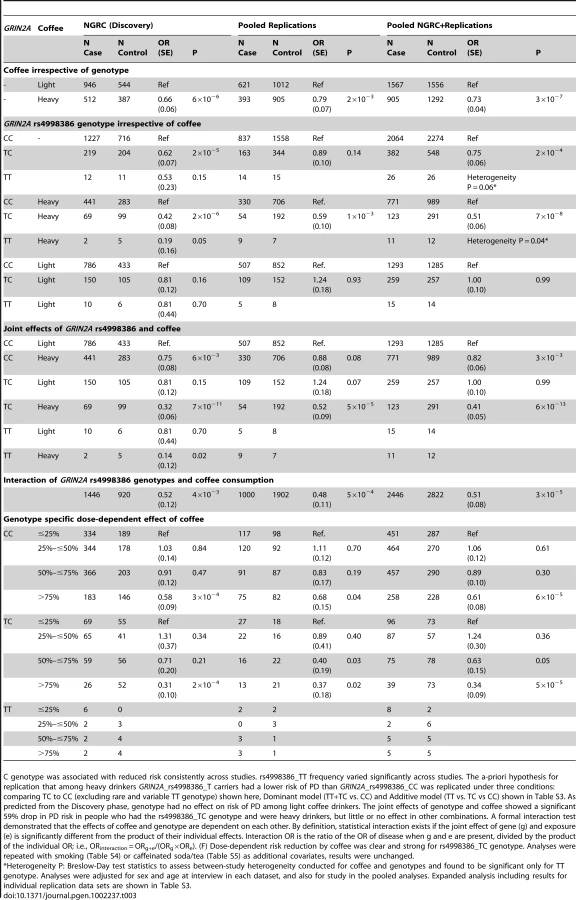
Hypothesis for replication
We used GWAIS as a means to identify genes that might enhance the inverse association of coffee with PD with the goal of carrying the discovery forward as a genetic marker for use in pharmacogenetic studies. Hence, the replication hypothesis was specified a-priori, based on results of NGRC, as follows: “Among heavy coffee drinkers, carriers of rs4998386_T allele have lower risk of PD than carriers of rs4998386_CC genotype”. Although this test does not reflect our most significant results, it is the test that has the clearest interpretation because it keeps the effect of coffee constant. For example, comparing TC+heavy vs. CC+light gave larger effect size and the P value was 3-orders of magnitude lower than the specified hypothesis, however, unlike our hypothesis, the test included coffee, which would have made it difficult to draw firm conclusions about the effect of genotype on coffee's inverse association with PD.
Potential confounders
Before attempting replication, the following analyses were conducted to identify potential confounders (Table S2). We tested the frequency of rs4998386 and coffee use across disease-specific strata and population structure. There was no evidence for heterogeneity by presence or absence of family history of PD, age at onset, or recruitment site. rs4998386 frequency was different between Ashkenazi-Jewish and non-Jewish individuals (P = 0.02) and across the European countries of ancestral origin (P = 3×10−3) in cases, but not in controls, which, PD being heterogeneous, may indicate different ethnic-specific clusters of disease subtypes as has been noted for LRRK2-associated PD [34]. Not surprisingly, heavy coffee use was associated with smoking (P<10−4), which itself is inversely associated with PD risk independently of coffee [12]. Adjusting for smoking, in addition to other covariates, did not change the results (Table S4). We also repeated the analyses adjusting for caffeinated soda and caffeinated tea consumption and found the results to be robust (Table S5). Some reports suggest persons with PD are more likely to avoid sensation seeking and addictive behaviors [35] and GRIN2A polymorphisms have been implicated in predisposition to heroin addiction [36] and smoking [37] raising the concern that our results could have been confounded if the GRIN2A SNPs identified here were associated with habitual coffee drinking. However, there was no evidence for association between any of the GRIN2A SNPs and heavy vs. light coffee consumption in cases and controls combined (OR = 0.95–1.01, P = 0.61–0.94).
Replication
See Table 3, Table S3. The a-priori hypothesis for replication that among heavy drinkers GRIN2A_rs4998386_T carriers had a lower risk of PD than GRIN2A_rs4998386_CC was replicated comparing TC to CC (excluding rare heterogeneous TT genotype): OR = 0.59, P = 10−3; under Additive model (TT vs. TC vs. CC): OR = 0.77, P = 0.04; and Dominant model (TT+TC vs. CC): OR = 0.70, P = 0.01. Note that the Additive and Dominant models included the TT genotype which is rare and its frequency varied significantly across datasets (Table 3, Table S6). The TC vs. CC comparison is more robust for this reason; Additive and Dominant model are shown for completeness. As seen in NGRC data, genotype had no effect on risk of PD among light coffee drinkers in Replication or combined data (OR = 1.0, P = 0.99).
In pooled Replication (without Discovery), the [SNP+SNP*coffee] joint test yielded P2df = 2.3×10−3 comparing TC to CC (excluding rare heterogeneous TT genotype); P2df = 0.12 for the Additive model, P2df = 0.02 for the Dominant model. The pooled analysis of Replication and Discovery with the [SNP+SNP*coffee] joint test yielded, P2df = 1.9×10−7 comparing TC to CC (excluding rare heterogeneous TT genotype), P2df = 1.4×10−5 for the Additive model, and P2df = 8.6×10−7 for the Dominant model.
In pooled data, compared to the light coffee drinkers with GRIN2A_rs4998386_CC genotype (the group with highest risk), heavy coffee use (with CC genotype) reduced risk by 18% (OR = 0.82, P = 3×10−3), having GRIN2A_rs4998386_T allele (light coffee) had no effect on risk (OR = 1.0, P = 0.99), but the combination of heavy coffee use and GRIN2A_rs4998386_TC genotype was associated with a highly significant 59% risk reduction (OR = 0.41, P = 6×10−13) (Table 3, Joint effects of GRIN2A rs4998386 and coffee).
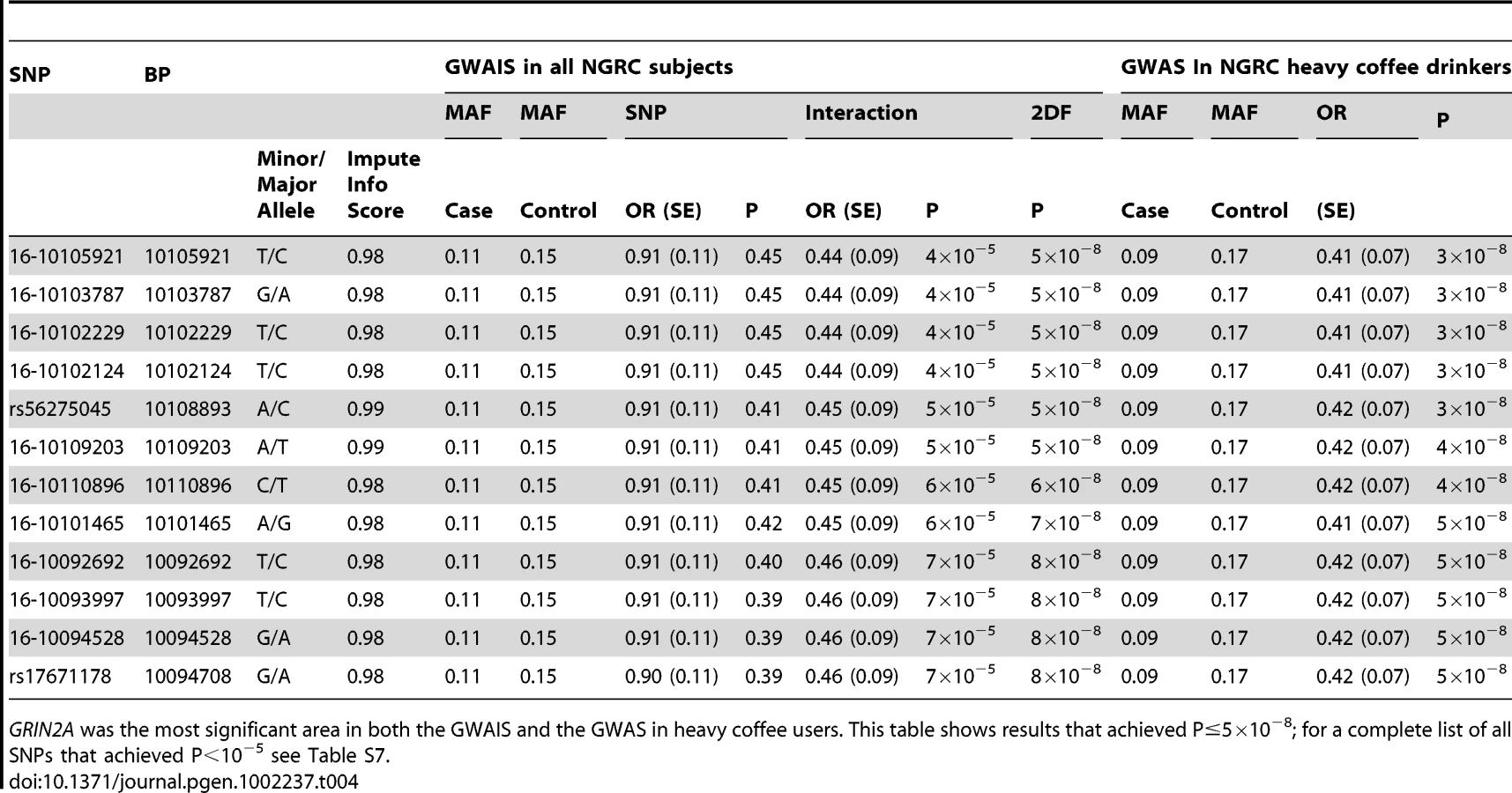
Imputation
See Table 4, Table S7. The array used in the study, Illumina OMNI-1 had nearly a million SNPs, which is a relatively dense coverage, but which could be further improved by imputing the SNPs that were not on the array using 1000 Genomes and HapMap data, a practice that has successfully aided many projects. After QC, we had over 3.5 million imputed and genotyped SNPs per individual in NGRC, each with information score ≥0.95 (measure of imputation certainty), and each passing standard GWAS QC. Imputation could only be applied to NGRC (Discovery) because only NGRC had genome-wide data. GWAIS and GWAS analysis of the GRIN2A region with imputed SNPs uncovered a block of densely linked SNPs embedded amongst the genotyped GRIN2A, six of which achieved P2df≤5×10−8 in GWAIS (Table 4). The interaction term was OR = 0.44, P = 4×10−5 (Table 4). In GWAS conducted in heavy coffee drinkers, 12 SNPs achieved P = 3×10−8 to 5×10−8 with OR = 0.41–0.42 (Table 4).
Discussion
In a genome-wide gene-environment study we identified GRIN2A as a genetic modifier of the inverse association of coffee with the risk of developing PD. The discovery was made in NGRC, and replicated in independent data. Risk reduction by heavy coffee use, which was estimated to be 27% on average, was genotype-specific and varied according to GRIN2A genotype from 18% (P = 3×10−3) for individuals with rs4998386_CC genotype to 59% (P = 6×10−13) for those with rs4998386_TC genotype. When coffee intake was categorized in four doses, the dose trend was more prominent in individuals with rs4998386_T allele than those with rs4998386_CC genotype, with the 3rd and 4th quartiles exhibiting only 11% and 39% risk reduction for rs4998386_CC carriers, vs. 37% and 66% for rs4998386_T carriers. With imputation we uncovered a block of GRIN2A SNPs not included on the genotyping array, which achieved P = 3×10−8 to 5×10−8. We propose GRIN2A as a new modifier gene for PD, and posit that if coffee-consumption is considered, GRIN2A may prove to be one of the most important PD-associated genes to have emerged from genome-wide studies. We base this suggestion on statistics, biology and the potential for immediate translation to clinical medicine, as we discuss below.
GRIN2A had not previously been tested as a candidate gene for PD, and was not detected in PD GWAS which have all been examining gene main effects without considering interactions with relevant environmental exposures. The most significant and consistently replicated main effects detected to date are for SNCA, MAPT and HLA. Here we added, for the first time, a common and relevant environmental exposure (coffee) to a genome-wide study. Inclusion of coffee allowed GRIN2A to rise to the top. In the gene-environment (GWAIS) model, GRIN2A surpassed SNCA, MAPT and HLA in statistical significance. Among heavy coffee drinkers, the impact of GRIN2A on PD risk (measured as OR) was 50% greater, and 2 to 5 orders of magnitude more significant (measured as P value) than the strongest associations reported for SNCA, MAPT or HLA. This study is proof of concept that inclusion of environmental factors can help identify disease-associated genes that are missed in SNP-only GWAS.
GRIN2A is an important gene for the central nervous system. Accelerated evolution of GRIN2A in primates is said to have contributed to the dramatic increase in the size and complexity of the human brain which defines human evolution [38]. GRIN2A encodes a subunit of the N-methyl-D-aspartate-2A (NMDA) glutamate receptor. It is central to excitatory neurotransmission and the control of movement and behavior [39]–[41]. The literature suggest imbalances in NMDA-dependent neurotransmission contribute to neurodegeneration in PD, possibly through massive influx of calcium and impaired mitochondrial function leading to apoptosis; and/or disruption of glutamate-mediated autophagy which is implicated in degradation and removal of proteins like α-synuclein (see [42] for review). The portion of intron 3 containing SNPs with the most significant associations (from base pair 9978046 to base pair 10128367, Table 1, Table 2, and Table 4) includes numerous transcription factor binding sites and two peaks of enhanced histone H3K4 mono-methylation (http://genome.ucsc.edu) [43]. Polymorphisms throughout this region could therefore disrupt regulatory elements, potentially leading to variation in levels of GRIN2A transcript. GRIN2A is expressed at high levels in the brain, most notably in the subthalamic nucleus (STN) [44]. Pharmacologic inhibition of STN with an NMDA-antagonist reduces nigral neuron loss in a rodent model of PD [45]. Deep-brain-stimulation, which also targets STN, is an effective surgical symptomatic therapy for PD.
The other piece of this finding is coffee/caffeine. Our study was not designed to distinguish the active ingredients in coffee. However, we note that the largest replication study (PAGE) measured specifically the caffeine intake in mg from all food sources (drink, food, and chocolate) and replicated our hypothesis and interaction robustly. We also found trends for inverse association of tea and soda with PD, and interestingly, the varied effect size and strength of association was consistent with the relative amount of caffeine in each drink (Table S5). Thus, our data are consistent with experimental observations that caffeine is neuroprotective. Caffeine is an adenosine A2A-receptor antagonist. A2A-receptor enhances calcium influx via NMDA [41] and A2A-receptor antagonists are neuroprotective in animal models of PD; they attenuate excitotoxicity by reducing extracellular glutamate levels in the striatum [46], [47]. Thus interaction between coffee/caffeine and GRIN2A is biologically plausible, and can help formulate testable hypotheses towards a better understanding of the disease pathogenesis.
GRIN2A genotyping may be useful for pharmacogenetic studies. Genetics has not yet entered drug development for PD but the time is here. We now have several susceptibility loci (SNCA, MAPT, HLA, BST1, PARK16, GAK [5]–[10]) that can help identify individuals who are at moderately increased risk of developing PD. We also have at least one neuroprotective compound (coffee/caffeine) which can be pharmacologically modified to alleviate its undesirable side effects. GRIN2A genotyping might also inform treatments for people who already have PD. L-DOPA, the primary PD drug for 40 years, does not slow disease progression and has serious side effects. Clinical trials for new PD drugs have not found drugs that surpass the symptomatic benefits of L-DOPA. There have been numerous drug trials for glutamate-receptor blockers as well as for selective A2A-receptor antagonists. Most were shown to be safe, well tolerated and beneficial [17], [18], [48]; however, the majority did not reach the regulatory threshold for efficacy to be approved as PD drugs. We wonder if some of these clinical trials will succeed if patients are subdivided by GRIN2A genotype. We acknowledge the distinction that the present study examined risk of developing PD; whereas clinical trials have thus far aimed for symptomatic improvements in patients. Nonetheless, there are sufficient parallels to suggest that GRIN2A genotype might also influence efficacy of glutamate-receptor antagonists and A2A-receptor antagonists. This is a simple and inexpensive hypothesis that can be tested in future, ongoing and even closed clinical trials that have banked DNA.
Common non-coding variants in GRIN2A have been associated with Huntington disease (HD) [49], [50] and schizophrenia [51], and rare mutations have been described in patients with neurodevelopmental phenotypes [52]. Schizophrenia is associated with a (GT)n repeat in the GRIN2A promoter that may increase disease risk by suppressing gene expression [51]. Three GRIN2A SNPs have been associated with onset-age of HD; they are conserved and reportedly tag a binding site for CCAAT/enhancer-binding protein [49], [50]. HD and PD are both neurodegenerative movement disorders, thus the possibility of a common genetic element was of interest. The reported HD-associated GRIN2A SNPs, rs1969060, rs8057394 and rs2650427, were not on the genotyping array but were imputed with high fidelity (information score >0.99). They map within the 150 kb region identified here for PD, they are in strong LD with PD-associated SNPs defined by D' (0.48–1.0) but not by r2 (0–0.33) (Figure S4). We tested the HD SNPs for association with onset age and risk of PD in NGRC while conditioning on the neighboring top PD SNP (rs4998386). One HD SNP, rs8057394, yielded OR = 0.85, P = 0.02 for PD overall; OR = 0.79, P = 0.04 for heavy coffee drinkers; and OR = 0.90, P = 0.24 for light coffee drinkers. We found no other evidence for association of HD SNPs with PD, including when we jointly tested HD SNPs and possible interaction with coffee [SNP+SNP*coffee] on risk or onset of PD. Conversely, we retested, in NGRC, the association of top genotyped PD SNP (rs4998386) with PD, conditioning on HD SNP (rs8057394) and found it to be robust (P2df = 8×10−6).
Unlike GWAS, which is now a fully standardized practice, there is no established protocol for testing gene*environment interaction on a whole-genome scale. Our strategy of starting with the joint test (GWAIS) and following up with GWAS in subgroups stratified by exposure was driven by the aims of our study. In Table S7 we present a side-by-side comparison of the results for the top GRIN2A SNPs (P<10−5), when analyzed for main effect (GWAS), for interaction, with Kraft's joint test, and in GWAS stratified by exposure. Amassing a large enough sample size for GWAIS is challenging. GWAIS requires larger sample sizes than GWAS yet there exist fewer samples that have data on relevant environmental exposures in addition to DNA and phenotype. To our knowledge, NGRC is the largest genetic study of PD that has collected exposure data. No other publically available PD GWAS has coffee data, eliminating the possibility of in-silico replication. We were able to identify and get access to only 3 datasets that had DNA and coffee, giving us a total sample size of 393 cases and 905 controls to replicate the GRIN2A effect in heavy coffee drinkers. In contrast, replications and meta-analyses for gene-only GWAS now have over 17,000 PD cases and controls [10]. We detected the known and confirmed PD-associated genes (SNCA, MAPT and HLA) in GWAIS but at much lower significance levels than in GWAS because of the smaller sample size with coffee data and the added degree of freedom in GWAIS. It is noteworthy, however, that at P2df = 10−6, GRIN2A surpassed all known PD loci in significance. With the aid of imputation, we achieved P = 3×10−8 for a 2.4-fold difference in genotype specific effect of coffee on risk of PD. Importantly, we were able to replicate the hypothesis that we set out a-priori based on discovery.
Supporting Information
Zdroje
1. ManolioTACollinsFSCoxNJGoldsteinDBHindorffLA 2009 Finding the missing heritability of complex diseases. Nature 461 747 753
2. ThomasD 2010 Gene–environment-wide association studies: emerging approaches. Nat Rev Genet 11 259 272
3. DorseyERConstantinescuRThompsonJPBiglanKMHollowayRG 2007 Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 68 384 386
4. HardyJ 2010 Genetic analysis of pathways to Parkinson disease. Neuron 68 201 206
5. HamzaTHZabetianCPTenesaALaederachAMontimurroJ 2010 Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson's disease. Nat Genet 42 781 785
6. SatakeWNakabayashiYMizutaIHirotaYItoC 2009 Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat Genet 41 1303 1307
7. Simon-SanchezJSchulteCBrasJMSharmaMGibbsJR 2009 Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet 41 1308 1312
8. PankratzNWilkJBLatourelleJCDeStefanoALHalterC 2009 Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet 124 593 605
9. EdwardsTLScottWKAlmonteCBurtAPowellEH 2010 Genome-Wide Association Study Confirms SNPs in SNCA and the MAPT Region as Common Risk Factors for Parkinson Disease. Ann Hum Genet 74 97 109
10. NallsMAPlagnolVHernandezDGSharmaMSheerinUM 2011 Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet 377 641 649
11. HernanMATakkoucheBCaamano-IsornaFGestal-OteroJJ 2002 A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson's disease. Ann Neurol 52 276 284
12. PowersKKayDFactorSZabetianCHigginsD 2008 Combined effects of smoking, coffee and NSAIDs on Parkinson's disease risk. Mov Disord 23 88 95
13. CostelloSCockburnMBronsteinJZhangXRitzB 2009 Parkinson's disease and residential exposure to maneb and paraquat from agricultural applications in the central valley of California. Am J Epidemiol 169 919 926
14. McCullochCCKayDMFactorSASamiiANuttJG 2008 Exploring gene-environment interactions in Parkinson's disease. Hum Genet 123 257 265
15. KraftPYenYCStramDOMorrisonJGaudermanWJ 2007 Exploiting gene-environment interaction to detect genetic associations. Hum Hered 63 111 119
16. ChenJFXuKPetzerJPStaalRXuYH 2001 Neuroprotection by caffeine and A(2A) adenosine receptor inactivation in a model of Parkinson's disease. J Neurosci 21 RC143
17. LeWittPAGuttmanMTetrudJWTuitePJMoriA 2008 Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces “off” time in Parkinson's disease: a double-blind, randomized, multicenter clinical trial (6002-US-005). Ann Neurol 63 295 302
18. FernandezHHGreeleyDRZweigRMWojcieszekJMoriA 2010 Istradefylline as monotherapy for Parkinson disease: results of the 6002-US-051 trial. Parkinsonism Relat Disord 16 16 20
19. Simon-SanchezJScholzSMatarin MdelMFungHCHernandezD 2008 Genomewide SNP assay reveals mutations underlying Parkinson disease. Hum Mutat 29 315 322
20. RitzBRManthripragadaADCostelloSLincolnSJFarrerMJ 2009 Dopamine transporter genetic variants and pesticides in Parkinson's disease. Environ Health Perspect 117 964 969
21. ChenHHuangXGuoXMailmanRBParkY 2010 Smoking duration, intensity, and risk of Parkinson disease. Neurology 74 878 884
22. HughesAJDanielSEBen-ShlomoYLeesAJ 2002 The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain 125 861 870
23. HancockDBMartinERStajichJMJewettRStacyMA 2007 Smoking, caffeine, and nonsteroidal anti-inflammatory drugs in families with Parkinson disease. Arch Neurol 64 576 580
24. ThompsonFEKipnisVMidthuneDFreedmanLSCarrollRJ 2008 Performance of a food-frequency questionnaire in the US NIH-AARP (National Institutes of Health-American Association of Retired Persons) Diet and Health Study. Public Health Nutr 11 183 195
25. ManningAKLaValleyMLiuCTRiceKAnP 2010 Meta-analysis of gene-environment interaction: joint estimation of SNP and SNP×environment regression coefficients. Genet Epidemiol 35 11 18
26. AschardHHancockDBLondonSJKraftP 2011 Genome-wide meta-analysis of joint tests for genetic and gene-environment interaction effects. Hum Hered 70 292 300
27. PurcellSNealeBTodd-BrownKThomasLFerreiraMA 2007 PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81 559 575
28. NeterJKunterMNachtsheimCWassermanW 1996 Applied Linear Statistical Models. 52 53
29. GaoXBeckerLCBeckerDMStarmerJDProvinceMA 2010 Avoiding the high Bonferroni penalty in genome-wide association studies. Genet Epidemiol 34 100 105
30. HowieBNDonnellyPMarchiniJ 2009 A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5 e1000529 doi:10.1371/journal.pgen.1000529
31. BarrettJCFryBMallerJDalyMJ 2005 Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21 263 265
32. LinDYHuangBE 2007 The use of inferred haplotypes in downstream analyses. Am J Hum Genet 80 577 579
33. WangKLiMHadleyDLiuRGlessnerJ 2007 PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 17 1665 1674
34. ZabetianCPHutterCMYearoutDLopezANFactorSA 2006 LRRK2 G2019S in families with Parkinson disease who originated from Europe and the Middle East: evidence of two distinct founding events beginning two millennia ago. Am J Hum Genet 79 752 758
35. EvansAHLawrenceADPottsJMacGregorLKatzenschlagerR 2006 Relationship between impulsive sensation seeking traits, smoking, alcohol and caffeine intake, and Parkinson's disease. J Neurol Neurosurg Psychiatry 77 317 321
36. LevranOLondonoDO'HaraKRandesiMRotrosenJ 2009 Heroin addiction in African Americans: a hypothesis-driven association study. Genes Brain Behav 8 531 540
37. VinkJMSmitABde GeusEJSullivanPWillemsenG 2009 Genome-wide association study of smoking initiation and current smoking. Am J Hum Genet 84 367 379
38. DorusSVallenderEJEvansPDAndersonJRGilbertSL 2004 Accelerated evolution of nervous system genes in the origin of Homo sapiens. Cell 119 1027 1040
39. ShenWFlajoletMGreengardPSurmeierDJ 2008 Dichotomous dopaminergic control of striatal synaptic plasticity. Science 321 848 851
40. JinXCostaRM 2010 Start/stop signals emerge in nigrostriatal circuits during sequence learning. Nature 466 457 462
41. HigleyMJSabatiniBL 2010 Competitive regulation of synaptic Ca2+ influx by D2 dopamine and A2A adenosine receptors. Nat Neurosci 13 958 966
42. CaudleWMZhangJ 2009 Glutamate, excitotoxicity, and programmed cell death in Parkinson disease. Exp Neurol 220 230 233
43. BirneyEStamatoyannopoulosJADuttaAGuigoRGingerasTR 2007 Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447 799 816
44. SuAIWiltshireTBatalovSLappHChingKA 2004 A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A 101 6062 6067
45. BlandiniFNappiGGreenamyreJT 2001 Subthalamic infusion of an NMDA antagonist prevents basal ganglia metabolic changes and nigral degeneration in a rodent model of Parkinson's disease. Ann Neurol 49 525 529
46. SchwarzschildMAXuKOztasEPetzerJPCastagnoliK 2003 Neuroprotection by caffeine and more specific A2A receptor antagonists in animal models of Parkinson's disease. Neurology 61 S55 61
47. TebanoMTPintorAFrankCDomeniciMRMartireA 2004 Adenosine A2A receptor blockade differentially influences excitotoxic mechanisms at pre- and postsynaptic sites in the rat striatum. J Neurosci Res 77 100 107
48. ShoulsonIPenneyJMcDermottMSchwidSKaysonE 2001 A randomized, controlled trial of remacemide for motor fluctuations in Parkinson's disease. Neurology 56 455 462
49. ArningLSaftCWieczorekSAndrichJKrausPH 2007 NR2A and NR2B receptor gene variations modify age at onset in Huntington disease in a sex-specific manner. Hum Genet 122 175 182
50. AndresenJMGayanJChernySSBrocklebankDAlkorta-AranburuG 2007 Replication of twelve association studies for Huntington's disease residual age of onset in large Venezuelan kindreds. J Med Genet 44 44 50
51. ItokawaMYamadaKYoshitsuguKToyotaTSugaT 2003 A microsatellite repeat in the promoter of the N-methyl-D-aspartate receptor 2A subunit (GRIN2A) gene suppresses transcriptional activity and correlates with chronic outcome in schizophrenia. Pharmacogenetics 13 271 278
52. EndeleSRosenbergerGGeiderKPoppBTamerC Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet 42 1021 1026
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 8
Nejčtenější v tomto čísle
- An EMT–Driven Alternative Splicing Program Occurs in Human Breast Cancer and Modulates Cellular Phenotype
- Chromosome Painting Reveals Asynaptic Full Alignment of Homologs and HIM-8–Dependent Remodeling of Chromosome Territories during Meiosis
- Discovery of Sexual Dimorphisms in Metabolic and Genetic Biomarkers
- Regulation of p53/CEP-1–Dependent Germ Cell Apoptosis by Ras/MAPK Signaling