
Genetic Interactions Involving Five or More Genes Contribute to a Complex Trait in Yeast
Although it is well known that interactions among genetic variants contribute to many complex traits, the forms of these interactions have not been fully characterized. Most work on this problem to date has focused on relatively simple cases involving two or three loci. However, higher-order interactions involving larger numbers of loci can also occur, and may have significant effects on the relationship between genotype and phenotype. In this paper, we dissect a colony morphology trait that segregates in a cross of two yeast strains and is caused by genetic interactions among five or more loci. Our work demonstrates that higher-order interactions can have major phenotypic effects, and provides novel insights into the genetic and molecular basis of these interactions.
Published in the journal:
. PLoS Genet 10(5): e32767. doi:10.1371/journal.pgen.1004324
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004324
Summary
Although it is well known that interactions among genetic variants contribute to many complex traits, the forms of these interactions have not been fully characterized. Most work on this problem to date has focused on relatively simple cases involving two or three loci. However, higher-order interactions involving larger numbers of loci can also occur, and may have significant effects on the relationship between genotype and phenotype. In this paper, we dissect a colony morphology trait that segregates in a cross of two yeast strains and is caused by genetic interactions among five or more loci. Our work demonstrates that higher-order interactions can have major phenotypic effects, and provides novel insights into the genetic and molecular basis of these interactions.
Introduction
Understanding the genetic basis of complex traits is critical for advancing medicine, evolutionary biology, and agriculture [1], [2]. A challenge to progress in this area is that genetic variants can interact, resulting in unexpected phenotypic consequences [3]–[7]. Most of our knowledge about these genetic interactions in natural systems comes from studies focused on two-locus interactions where at least one of the loci exhibits a measurable effect on its own (e.g., [8]). However, evidence suggests that genetic interactions involving three or more loci also occur [9], [10], and that loci participating in such interactions may not individually have detectable effects [11]. Determining how these higher-order interactions arise and influence phenotypic variation could help solve the ‘missing heritability’ problem faced by geneticists studying humans and model species [12].
In this paper, we describe the genetic basis of a complex trait that is influenced by higher-order interactions. We identified this phenotype, a dramatic change in the morphology of Saccharomyces cerevisiae colonies, in a cross of haploid derivatives of the lab strain BY4716 and the clinical isolate 322134S (hereafter ‘BY’ and ‘3S’, respectively). The colony morphology trait in the BY×3S cross is similar to phenotypes described in other yeast isolates and crosses (e.g., [13]–[19]). Thus, by comprehensively determining the genetic basis of colony morphology variation among BY×3S offspring, we not only generate novel insights into how higher-order interactions contribute to phenotypic variation, but also provide new information regarding the genetic basis of a frequently studied model complex trait.
Results and Discussion
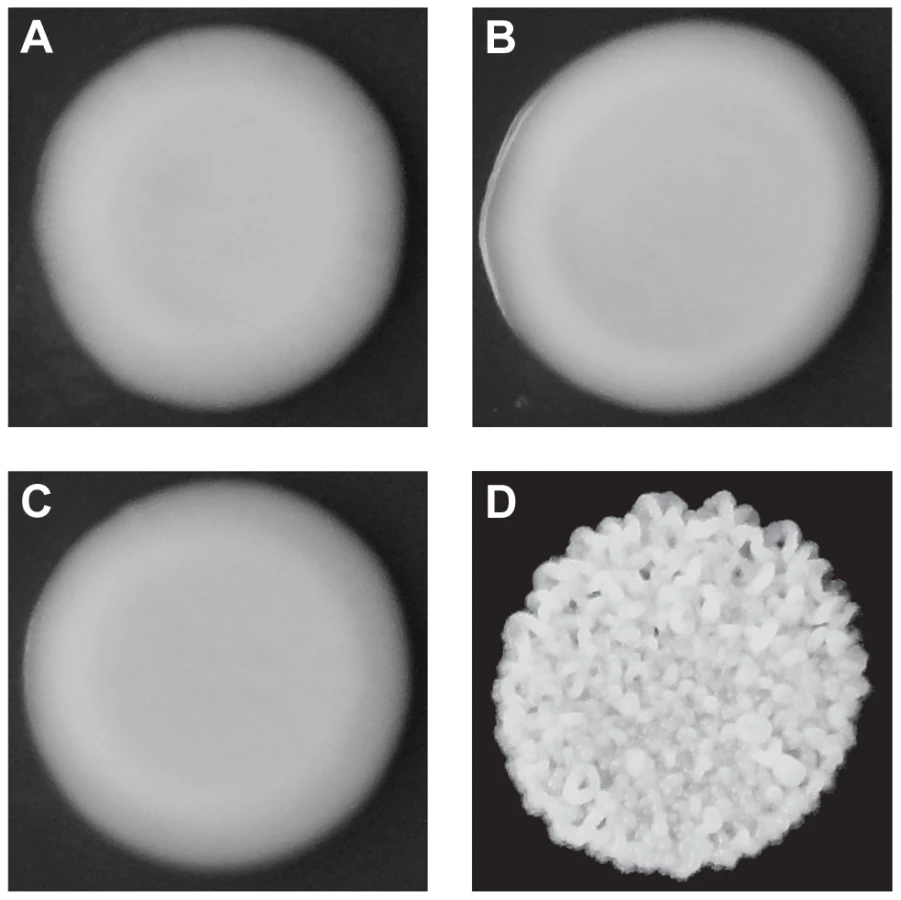
Although both BY and 3S, as well as most of their haploid offspring, form smooth colonies (Figure 1A–C), ∼2% of their progeny exhibited rough colonies when we examined 250 segregants (Figure 1D). Previous work has shown that such heritable variation in colony morphology in S. cerevisiae can arise due to naturally occurring polymorphisms or spontaneous mutations at chromosomal loci [13], [14], [18], [19], aneuploidies [17], and prions [15]. Unlike chromosomal loci, which should show stable inheritance across generations, aneuploidies and prions can be gained or lost, resulting in phenotypic switching. Multiple lines of evidence suggest that chromosomal loci are the primary cause of rough morphology in the BY×3S cross. Neither BY nor 3S exhibits rough morphology, indicating that the phenotype likely requires a combination of alleles from both of these strains. Consistent with this statement, we found that the frequency of rough morphology increased to 12.5% and 21.2% among recombinant haploid progeny obtained by backcrossing a rough segregant to BY and 3S, respectively (Tables S1 and S2; Methods). The higher frequency of rough segregants in backcrosses is expected if alleles from both parents contribute to the trait, as fewer causative alleles should segregate in the backcrosses than in the original cross. Further supporting the argument that our observations of rough morphology were due to chromosomal loci instead of transient factors, we found no evidence for chromosome-scale aneuploidies or phenotypic switching in the backcrossed segregant (Figure S1; Methods).
To identify loci that contribute to rough morphology, we generated thousands of random spores from the aforementioned backcrosses and used low-coverage whole genome sequencing to selectively genotype individuals that showed the phenotype (Methods). We obtained 92 and 88 rough segregants from the BY and 3S backcrosses, respectively. Using these data, we detected five genomic loci that were strongly enriched among these individuals but not among control segregants (Figure S2): three on Chromosomes IV, V, and XV inherited from 3S (Figure 2A), and two on Chromosomes XIII and XIV inherited from BY (Figure 2B). All of these loci, except the one on Chromosome XIV, were fixed among individuals with rough morphology.
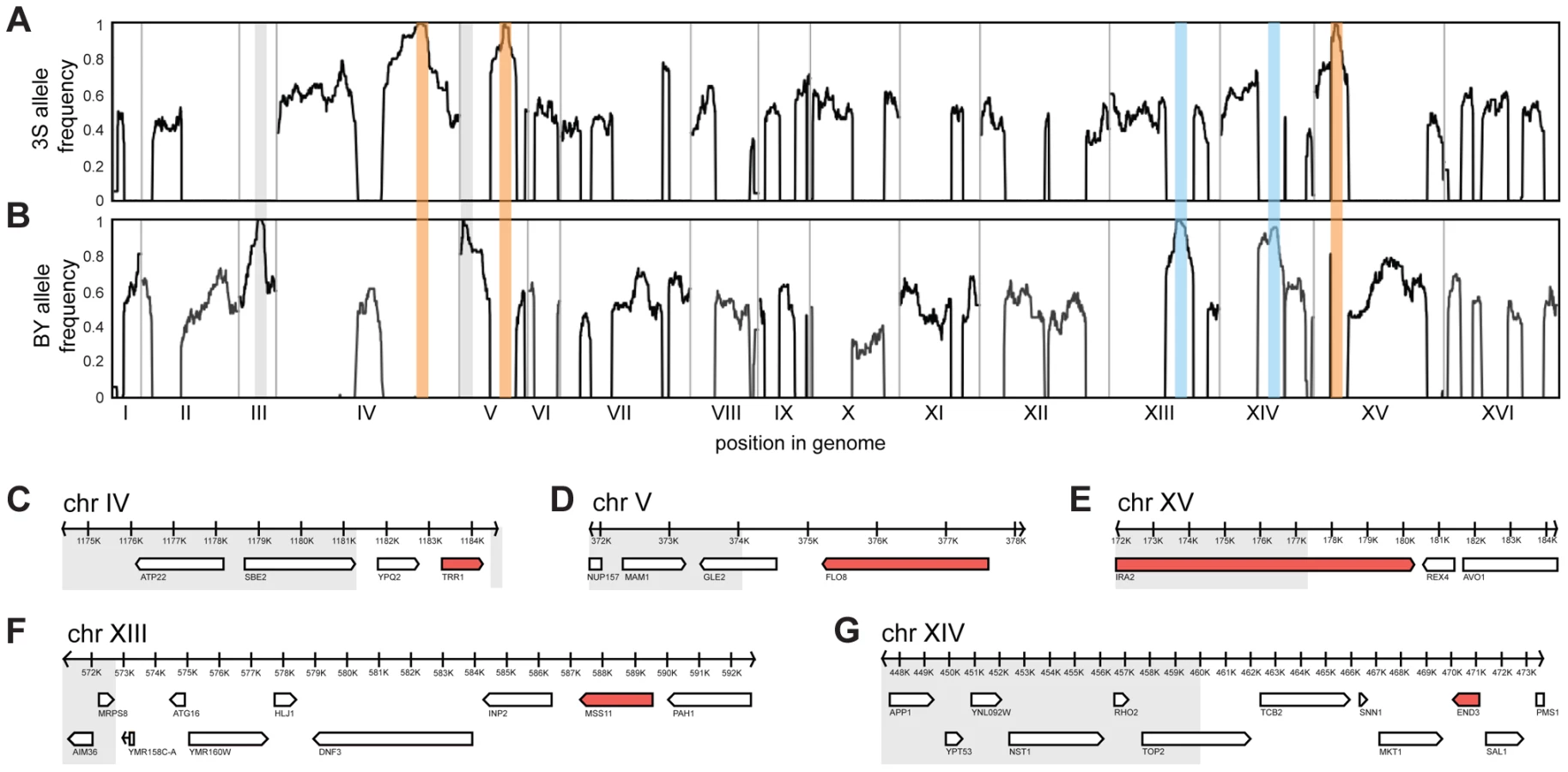
We attempted to determine causal genes underlying each of the five loci. Our initial resolution of the loci was between 4 and 14 genes (Figure 2C–G; Table S3; Methods). To decrease the number of candidate genes, we performed targeted genotyping on 19 additional backcross segregants, as well as 8 multi-locus introgression strains that had been subjected to 6 rounds of backcrossing with selection for the rough phenotype (Figure S3; Methods). This additional stage of genetic mapping refined the loci to between 2 and 9 genes per locus, and 22 genes in total (Figure 2C–G; Table S4, S5, S6). We deleted each of the 20 remaining non-essential candidate genes from one of the multi-locus introgression strains (Methods). Across these deletions, a single gene at each locus showed an effect on the phenotype: TRR1 (Chromosome IV), FLO8 (Chromosome V), MSS11 (Chromosome XIII), END3 (Chromosome XIV), and IRA2 (Chromosome XV) (Figure 2C–G). Because the two remaining candidate genes—AVO1 and TOP2—were essential, we examined them using an alternative strategy that suggested they do not contribute to the observed colony morphology variation (Text S1).
We used complementation tests to determine whether the five identified genes possess functional variation (Methods). Each haploid deletion strain was mated to three rough and three smooth haploid backcross progeny (Methods). These matings were designed to produce diploids that were homozygous for the required alleles at four of the causal loci and hemizygous for the fifth causal locus. For END3, FLO8, MSS11, and TRR1, the experiments provided support that the parental alleles differ in their effects. All matings of deletion strains to smooth backcross progeny produced smooth hemizygotes. Further, either two (in the cases of FLO8 and MSS11) or three (in the cases of TRR1 and END3) of the matings of deletion strains to rough backcross progeny produced rough hemizygotes (Figure 3A). However, for IRA2, the two possible hemizygotes showed no phenotypic difference, with both exhibiting smooth morphology (Figure 3A). IRA2 has been reported to show haploinsufficiency in growth rate experiments [20], and this haploinsufficiency may also explain some of our reciprocal hemizygosity results for this gene.
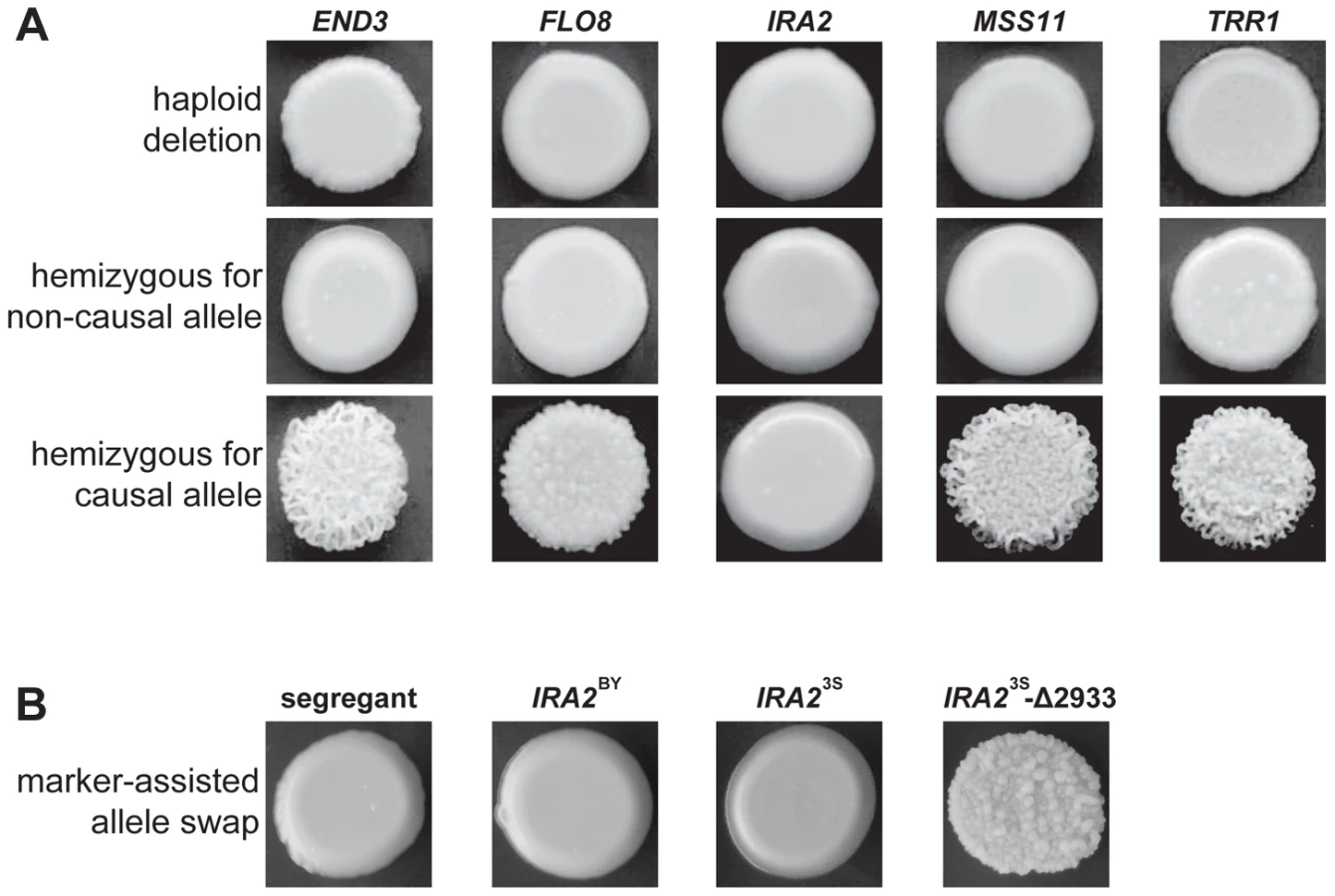
To provide stronger support for IRA2's role in the trait, we performed allele replacements of IRA2 in a smooth backcross segregant that carried the non-causal allele of IRA2, as well as the causal alleles of END3, FLO8, MSS11, and TRR1 (Methods). While transformations with the IRA23S allele had no phenotypic effect, we found that transformations with the IRA2 allele from the rough segregant that had been backcrossed resulted in a change from smooth to rough morphology (Figure 3B). Sequencing of IRA2 from 3S and the rough segregant revealed a single difference between the two alleles: a frameshift mutation that truncates the protein by 117 amino acids (hereafter referred to as IRA23S-Δ2933; Text S2). IRA2 is known to be hypermutable and spontaneous mutations in this gene have been shown to influence a variety of multicellular growth phenotypes [19], [21]. However, our results demonstrate that the effects of spontaneous mutations in IRA2 can depend on an individual's genotype at a number of additional genes. We also checked for IRA23S-Δ2933 in the four other rough individuals that we found in our original BY×3S mapping population. Three of these rough segregants possessed the frameshift mutation, suggesting that IRA23S-Δ2933 probably arose during the outgrowth of the BY/3S diploid prior to its sporulation.
Previous work by other groups identified functional polymorphisms in END3 and FLO8 that also segregate in our cross [22], [23]. BY has a premature stop mutation in FLO8 that prevents it from undergoing many forms of multicellular growth [22]. As for END3, a missense polymorphism in this gene contributes to variability in high temperature growth in a cross of the clinical isolate YJM789 and S288c, the progenitor of BY [23]. Of relevance to our study, this variant in END3 has effects that are strongly dependent on genetic background [24]. With respect to TRR1, the Saccharomyces Genome Resequencing Project [25] and our own sequencing data indicate that the BY and 3S alleles of this gene differ by a single nucleotide, which is a synonymous SNP in the 52nd codon of the gene: BY has an ATC codon and 3S has an ATT codon. Although both of these codons are recognized by the same isoleucine tRNA, the ATT codon is preferred by a nearly two-to-one ratio throughout the yeast genome, suggesting that the SNP might have an effect on translational efficiency. Only lab-derived S. cerevisiae strains carry the ATC allele that confers smooth morphology, while all other sequenced S. cerevisiae and S. paradoxus strains harbor the ATT allele that is likely involved in rough morphology. Work to determine the functional variant(s) in MSS11, which possesses a number of coding and noncoding polymorphisms that could have effects, is ongoing (Table S7).
The causal genes encode proteins with diverse cellular functions: End3 plays a role in clathrin-mediated endocytosis [26], [27], Flo8 and Mss11 are transcription factors that regulate cell-cell adhesion and multicellular phenotypes in S. cerevisiae [28], [29], Ira2 is a negative regulator of the RAS-cAMP pathway [30], and Trr1 is an enzyme involved in oxidative stress response [31], [32]. Flo8 and Mss11 physically interact [33], and IRA2 and MSS11 show a genetic interaction when both are knocked out [34]. To our knowledge, none of the other pairs of identified genes have been reported to interact at the biochemical, genetic, physical, or regulatory levels. To assess whether Flo8 and Mss11 might directly regulate the expression of the other genes, we examined existing data from calling card analyses, a technique that identifies genomic sites bound by transcription factors [16]. These results indicated that Flo8 and Mss11 are unlikely to bind the promoters of END3, IRA2, and TRR1, although admittedly the study involved a different strain than our cross parents.
After identifying causal genes at the five loci, we analyzed the effects of these genes in more detail by genotyping them in a panel of phenotyped segregants from dissected backcross tetrads (Methods). Every individual with rough morphology possessed the 3S allele of FLO8 and TRR1, the BY allele of MSS11, and IRA23S-Δ2933 (Figures 4A–B and S4A–B; Tables S8 and S9). Although most individuals with rough morphology carried END3BY, a small fraction of individuals with END33S also showed the trait (Figures 4B and S4C; Table S9), indicating that alleles at additional loci complement END33S.
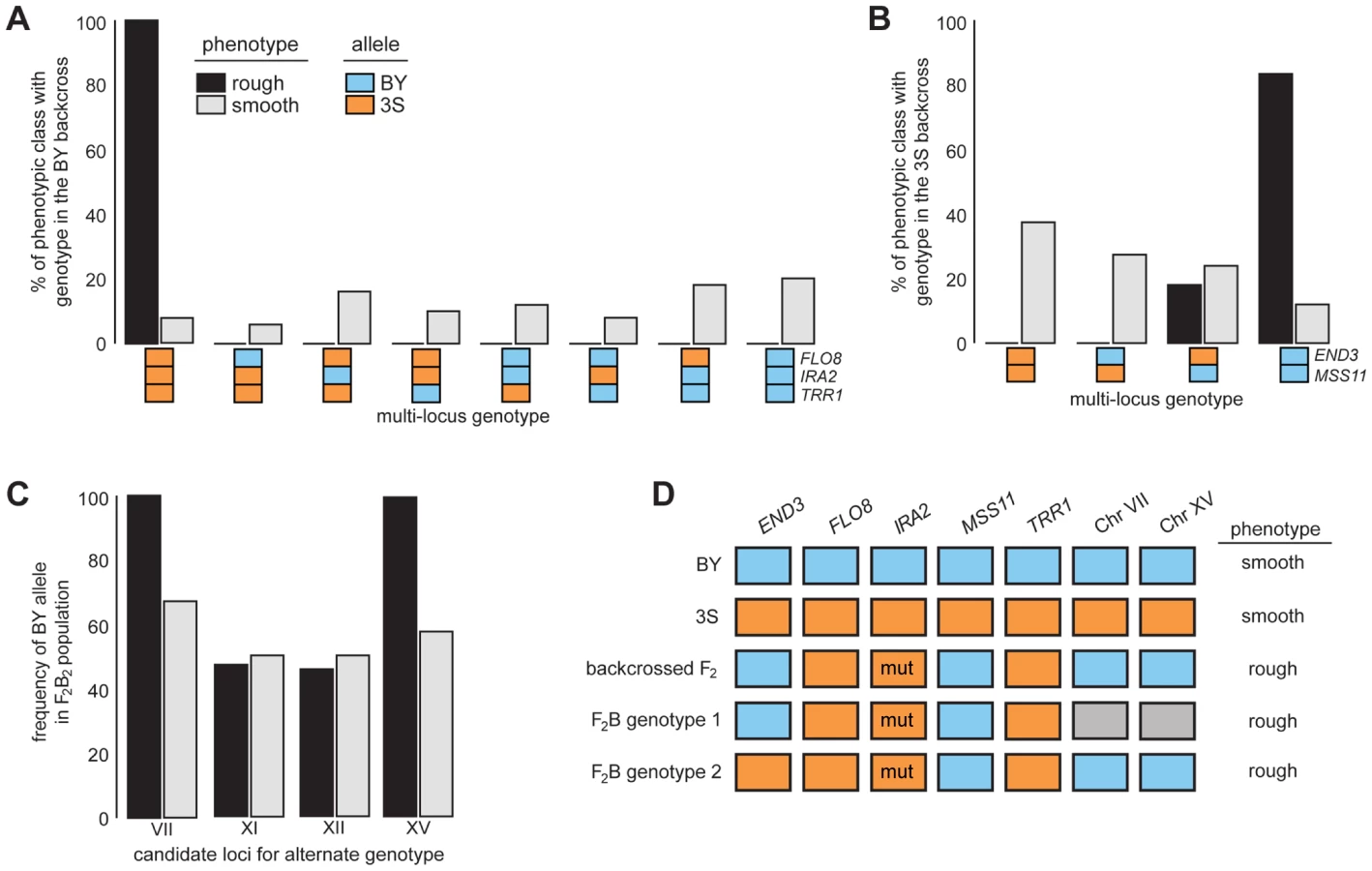
We more deeply investigated the genetic basis of rough morphology among individuals with END33S. First, we used a gene knockout strategy to check whether END33S is necessary for these individuals to exhibit rough morphology (Methods). end33SΔ strains were smooth (Figure S5), suggesting that the alternate genetic architecture for rough morphology requires END33S. Second, we tried to identify loci that complement END33S. Four rough END33S progeny were present in our sequenced mapping population from the 3S backcross. Among these segregants, we detected 11 previously unidentified genomic regions where individuals shared the same genotype (Figure S6; Table S10; Methods). We were able to reduce this set to four candidate loci on Chromosomes VII, XI, XII, and XV by genotyping additional backcross progeny (Table S11; Methods). To determine which of the four loci have causal roles in rough morphology, we mated a relevant backcross segregant to 3S and analyzed a panel of 51 second-generation backcross progeny (Table S12; Methods). The BY alleles at the Chromosome VII and XV loci were fixed among the 39 individuals with rough morphology, while the other two loci showed no evidence of playing a role in the trait (Figure 4C; Table S12). Given only individuals that carried BY alleles at both the Chromosome VII and XV loci exhibited rough morphology, it is likely that these loci genetically interact to complement END33S.
Our findings indicate that the segregant used for backcrossing carried more than one set of interacting alleles that can specify rough morphology (Figure 4D). Identifying the causal genes and genetic variants underlying the Chromosome VII and XV loci can thus shed light on how these different genotypes produce the same trait. However, our ongoing efforts to clone the causal factors at these loci are limited by the crude resolution of the present data (each locus is presently resolved to >60 kilobases; Table S10). We note that initial gene deletion experiments focused on 18 candidates (Table S13), including LAS17 and YAP1802, whose cognate proteins functionally interact with End3 [35], [36], have been unsuccessful. Moving forward, we plan to determine the genes that underlie the Chromosome VII and XV loci, and characterize their relationship with END3.
In summary, we have demonstrated that sets of five or more genetic variants can synthetically interact to produce major phenotypic effects. Alleles involved in these higher-order interactions may either be polymorphisms that segregate in natural populations or spontaneous mutations. Our results also illustrate that rather than functioning in a single biochemical pathway, protein complex, or regulatory circuit, the genes involved in higher-order interactions can play roles in a number of cellular processes. This finding implies that characterizing higher-order interactions using data from screens and annotations focused solely on reference genomes may be a challenge, and highlights how genetic variation can serve as a tool for detecting previously unidentified functional relationships among genes. Further, we have shown that multiple sets of alleles can interact to produce the same phenotypic effect. Additional work is necessary to determine how this latter finding is mediated at the molecular and systems levels. Overall, our study suggests that characterizing the larger-scale contribution of higher-order interactions to phenotypic variation is a necessary step in improving our basic understanding of the genotype-phenotype map.
Methods
Phenotyping of yeast colony morphology
All phenotyping experiments were performed on agar plates containing yeast extract and peptone (YP) with 2% ethanol as the carbon source (YPE). Prior to phenotyping, strains were grown up in liquid YP with 2% dextrose (YPD). Stationary-phase cultures were manually pinned onto YPE and allowed to grow for five days at 30°C, and were then imaged using a standard digital camera.
Assessing potential effects of transient heritable factors
Sequencing data from the rough segregant used in backcross experiments was examined at the chromosome-scale for evidence of aneuploidy. Average per base coverage of each chromosome was computed in R and compared to the genome-wide average. This segregant was also plated at low density on a large number of YPE plates. We screened tens of thousands of colonies for instances of phenotypic switching and observed no cases where an individual converted from rough to smooth morphology.
Generation of backcross segregants
Strains used in this paper contained the Synthetic Genetic Array marker system [37], which allowed us to easily generate large numbers of recombinant MATa progeny. All segregants discussed in the paper were MATa can1Δ::STE2pr-SpHIS5 his3Δ and all backcrosses involved mating these individuals to either a BY or a 3S strain that was MATα his3Δ. In these crosses, strains with opposite mating types were mixed together on a YPD plate and incubated for four hours at 30°C. Zygotes were then obtained by microdissection. To generate segregants, diploids were sporulated at room temperature using the protocol described by Guthrie and Fink [38]. Once sporulation had completed, spore cultures were digested with β-glucuronidase and then plated onto yeast nitrogen base (YNB) plates containing canavanine, as described previously [39]. Spores were plated at a density of roughly 100 to 200 colonies per plate.
Genome sequencing of backcross segregants
Whole genome sequencing libraries were prepared using the Illumina Nextera kit, with each of the backcross segregants barcoded with a unique sequence tag. The libraries were mixed together in equimolar fractions and sequenced on an Illumina HiSeq machine by the Beijing Genomics Institute using 100 base pair (bp) ×100 bp reads. Sequencing reads were then mapped to the S. cerevisiae reference genome using the Burrows-Wheeler Aligner (BWA) [40]. We used data from 36,756 high confidence SNPs that had been identified based on comparison of Illumina sequence data for 3S to the BY genome. Similar to Andolfatto et al. [41], we employed Hidden Markov Models (HMMs) to determine the haplotypes of the segregants based on the sequencing data. We computed the fraction of reads at each SNP that came from BY and used the vector of these fractions in HMMs that were implemented chromosome-by-chromosome in the HMM() package of the R statistical programming environment. Any segregants producing data that showed evidence of contamination, diploidy, or aneuploidy were excluded from genetic mapping and downstream analyses. Four and eight such individuals were left out of the BY and 3S mapping populations, respectively.
Genetic mapping
Genotypes inferred from the HMM were used in genetic mapping analyses. At each position in the genome, we determined the fraction of individuals that carried the allele from the parent not used in the backcross. We scanned the genome for alleles from the non-backcross parent that were detected in a large fraction of segregants. We report loci where these alleles were at 95% frequency or higher. To determine intervals in which causal genes were located, we identified the smallest region that was bounded by recombination breakpoints among individuals from a backcross that shared the same allele at a peak.
Generation and genotyping of dissected tetrads
Backcross diploids were sporulated and digested in β-glucuronidase to permit tetrad dissection. Standard microdissection techniques were used to isolate tetrads and separate individual spores.
Fine-mapping using multi-locus introgression strains and dissected backcross segregants
Haploid multi-locus introgression strains were constructed using six rounds of recurrent backcrossing with phenotypic selection, starting from the same segregant used in our backcross mapping experiment. Eight of these strains were generated, with four made by recurrently backcrossing to 3S and four made by recurrently backcrossing to BY. We also used a subset of individuals from the tetrad dissections that showed rough morphology. To conduct the fine-mapping, we typed these individuals at a number of markers in each interval using PCR and restriction digestion, or Sanger sequencing.
Genetic engineering experiments
All genes within causal loci were deleted using the CORE cassette, in the same manner described by Storici et al. [42]. Homology tails matching the 60 bases immediately up- and downstream of each gene were attached to the CORE cassette through PCR and introduced into cells using the Lithium Acetate method [43]. Selection for G418 resistance was used to screen for integration of the CORE cassette; correct integration was then checked using PCR. All deletions were performed in a haploid multi-locus introgression strain. To perform complementation tests, deletion strains were mated to multiple dissected segregants that carried either the causal or non-causal allele of the deleted gene, as well as the causal alleles at the four other involved genes. The same phenotyping methods described above were employed for these strains. To generate allele replacement strains for IRA2, a smooth segregant with the non-causal allele of IRA2 and the causal alleles at the other four loci was transformed using a modified form of adaptamer mediated allele replacement [44]. Transformations were conducted with two partially overlapping PCR products—a full-length amplicon of IRA2 that was tailed at the 3′ end with the 5′ portion of the kanMX cassette and a copy of the kanMX cassette that was tailed on the 3′ end with part of the intergenic region downstream of IRA2. Knock-ins were identified using selection on G418 and verified by Sanger sequencing.
Identification of loci that complement the 3S allele of END3
Sequenced strains from the backcross to 3S were partitioned based on their genotype at END3. We then screened these individuals for sites where they all carried BY alleles. A group of additional rough segregants with END33S that had been obtained during tetrad dissections were genotyped by PCR amplification and restriction digestion of markers across each of the new loci. One of these additional backcross segregants was mated to 3S, and a panel of rough progeny from this second-generation backcross were typed at the remaining candidate loci.
Supporting Information
Zdroje
1. Falconer DS, Mackay TF (1996) Introduction to quantitative genetics (4th edition). Harlow, England: Pearson Education Limited.
2. Lynch M, Walsh B (1998) Genetics and analysis of quantitative traits: Sinauer.
3. MackayTF (2014) Epistasis and quantitative traits: using model organisms to study gene-gene interactions. Nature reviews Genetics 15: 22–33.
4. PhillipsPC (2008) Epistasis–the essential role of gene interactions in the structure and evolution of genetic systems. Nat Rev Genet 9: 855–867.
5. LehnerB (2011) Molecular mechanisms of epistasis within and between genes. Trends Genet 27: 323–331.
6. HuangW, RichardsS, CarboneMA, ZhuD, AnholtRR, et al. (2012) Epistasis dominates the genetic architecture of Drosophila quantitative traits. Proceedings of the National Academy of Sciences of the United States of America 109: 15553–15559.
7. NelsonRM, PetterssonME, CarlborgO (2013) A century after Fisher: time for a new paradigm in quantitative genetics. Trends in genetics: TIG 29: 669–76 doi:10.1016/j.tig.2013.09.006
8. BremRB, StoreyJD, WhittleJ, KruglyakL (2005) Genetic interactions between polymorphisms that affect gene expression in yeast. Nature 436: 701–703.
9. DowellRD, RyanO, JansenA, CheungD, AgarwalaS, et al. (2010) Genotype to phenotype: a complex problem. Science 328: 469.
10. PetterssonM, BesnierF, SiegelPB, CarlborgO (2011) Replication and explorations of high-order epistasis using a large advanced intercross line pedigree. PLoS genetics 7: e1002180.
11. BloomJS, EhrenreichIM, LooWT, LiteTL, KruglyakL (2013) Finding the sources of missing heritability in a yeast cross. Nature 494: 234–237.
12. ManolioTA, CollinsFS, CoxNJ, GoldsteinDB, HindorffLA, et al. (2009) Finding the missing heritability of complex diseases. Nature 461: 747–753.
13. GranekJA, MagwenePM (2010) Environmental and genetic determinants of colony morphology in yeast. PLoS genetics 6: e1000823.
14. GranekJA, MurrayD, KayrkciO, MagwenePM (2013) The genetic architecture of biofilm formation in a clinical isolate of Saccharomyces cerevisiae. Genetics 193: 587–600.
15. HolmesDL, LancasterAK, LindquistS, HalfmannR (2013) Heritable remodeling of yeast multicellularity by an environmentally responsive prion. Cell 153: 153–165.
16. RyanO, ShapiroRS, KuratCF, MayhewD, BaryshnikovaA, et al. (2012) Global gene deletion analysis exploring yeast filamentous growth. Science 337: 1353–1356.
17. TanZ, HaysM, CromieGA, JefferyEW, ScottAC, et al. (2013) Aneuploidy underlies a multicellular phenotypic switch. Proceedings of the National Academy of Sciences of the United States of America 110: 12367–12372.
18. WilkeningS, LinG, FritschES, TekkedilMM, AndersS, et al. (2013) An Evaluation of High-Throughput Approaches to QTL Mapping in Saccharomyces cerevisiae. Genetics 196-: 853–65.
19. HalmeA, BumgarnerS, StylesC, FinkGR (2004) Genetic and epigenetic regulation of the FLO gene family generates cell-surface variation in yeast. Cell 116: 405–415.
20. PirP, GutteridgeA, WuJ, RashB, KellDB, et al. (2012) The genetic control of growth rate: a systems biology study in yeast. BMC systems biology 6: 4.
21. RoopJI, BremRB (2013) Rare variants in hypermutable genes underlie common morphology and growth traits in wild Saccharomyces paradoxus. Genetics 195: 513–525.
22. LiuH, StylesCA, FinkGR (1996) Saccharomyces cerevisiae S288C has a mutation in FLO8, a gene required for filamentous growth. Genetics 144: 967–978.
23. SteinmetzLM, SinhaH, RichardsDR, SpiegelmanJI, OefnerPJ, et al. (2002) Dissecting the architecture of a quantitative trait locus in yeast. Nature 416: 326–330.
24. SinhaH, NicholsonBP, SteinmetzLM, McCuskerJH (2006) Complex genetic interactions in a quantitative trait locus. PLoS genetics 2: e13.
25. LitiG, CarterDM, MosesAM, WarringerJ, PartsL, et al. (2009) Population genomics of domestic and wild yeasts. Nature 458: 337–341.
26. BenedettiH, RathsS, CrausazF, RiezmanH (1994) The END3 gene encodes a protein that is required for the internalization step of endocytosis and for actin cytoskeleton organization in yeast. Mol Biol Cell 5: 1023–1037.
27. TangHY, XuJ, CaiM (2000) Pan1p, End3p, and S1a1p, three yeast proteins required for normal cortical actin cytoskeleton organization, associate with each other and play essential roles in cell wall morphogenesis. Mol Cell Biol 20: 12–25.
28. KobayashiO, SudaH, OhtaniT, SoneH (1996) Molecular cloning and analysis of the dominant flocculation gene FLO8 from Saccharomyces cerevisiae. Molecular & general genetics: MGG 251: 707–715.
29. GagianoM, BesterM, van DykD, FrankenJ, BauerFF, et al. (2003) Mss11p is a transcription factor regulating pseudohyphal differentiation, invasive growth and starch metabolism in Saccharomyces cerevisiae in response to nutrient availability. Molecular microbiology 47: 119–134.
30. TanakaK, NakafukuM, TamanoiF, KaziroY, MatsumotoK, et al. (1990) IRA2, a second gene of Saccharomyces cerevisiae that encodes a protein with a domain homologous to mammalian ras GTPase-activating protein. Molecular and cellular biology 10: 4303–4313.
31. PedrajasJR, KosmidouE, Miranda-VizueteA, GustafssonJA, WrightAP, et al. (1999) Identification and functional characterization of a novel mitochondrial thioredoxin system in Saccharomyces cerevisiae. J Biol Chem 274: 6366–6373.
32. RossSJ, FindlayVJ, MalakasiP, MorganBA (2000) Thioredoxin peroxidase is required for the transcriptional response to oxidative stress in budding yeast. Mol Biol Cell 11: 2631–2642.
33. KimTS, KimHY, YoonJH, KangHS (2004) Recruitment of the Swi/Snf complex by Ste12-Tec1 promotes Flo8-Mss11-mediated activation of STA1 expression. Molecular and cellular biology 24: 9542–9556.
34. HoppinsS, CollinsSR, Cassidy-StoneA, HummelE, DevayRM, et al. (2011) A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. The Journal of cell biology 195: 323–340.
35. TarassovK, MessierV, LandryCR, RadinovicS, Serna MolinaMM, et al. (2008) An in vivo map of the yeast protein interactome. Science 320: 1465–1470.
36. HowardJP, HuttonJL, OlsonJM, PayneGS (2002) Sla1p serves as the targeting signal recognition factor for NPFX(1,2)D-mediated endocytosis. The Journal of cell biology 157: 315–326.
37. TongAH, EvangelistaM, ParsonsAB, XuH, BaderGD, et al. (2001) Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294: 2364–2368.
38. GuthrieC, FinkGR (1991) Guide to Yeast Genetics and Molecular Biology. Meth Enzymol 194: 429–663.
39. EhrenreichIM, TorabiN, JiaY, KentJ, MartisS, et al. (2010) Dissection of genetically complex traits with extremely large pools of yeast segregants. Nature 464: 1039–1042.
40. LiH, DurbinR (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760.
41. AndolfattoP, DavisonD, ErezyilmazD, HuTT, MastJ, et al. (2011) Multiplexed shotgun genotyping for rapid and efficient genetic mapping. Genome research 21: 610–617.
42. StoriciF, LewisLK, ResnickMA (2001) In vivo site-directed mutagenesis using oligonucleotides. Nature biotechnology 19: 773–776.
43. GietzRD, WoodsRA (2002) Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods in enzymology 350: 87–96.
44. ErdenizN, MortensenUH, RothsteinR (1997) Cloning-free PCR-based allele replacement methods. Genome research 7: 1174–1183.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 5
Nejčtenější v tomto čísle
- PINK1-Parkin Pathway Activity Is Regulated by Degradation of PINK1 in the Mitochondrial Matrix
- Phosphorylation of a WRKY Transcription Factor by MAPKs Is Required for Pollen Development and Function in
- Null Mutation in PGAP1 Impairing Gpi-Anchor Maturation in Patients with Intellectual Disability and Encephalopathy
- p53 Requires the Stress Sensor USF1 to Direct Appropriate Cell Fate Decision